

Development of the Chromatographic Method for Simultaneous Determination of Azaperone and Azaperol in Animal Kidneys and Livers

Abstract
:1. Introduction
2. Results and Discussion
2.1. Sample Preparation
2.1.1. Optimization of the Ratio of Animal Tissue Mass to Acetonitrile Volume
2.1.2. Selection of the Lyophilization Temperature
2.1.3. Optimization of Ethanol Volume after Lyophilization
2.1.4. Optimization of Perchloric Acid Volume
2.2. Chromatographic Conditions
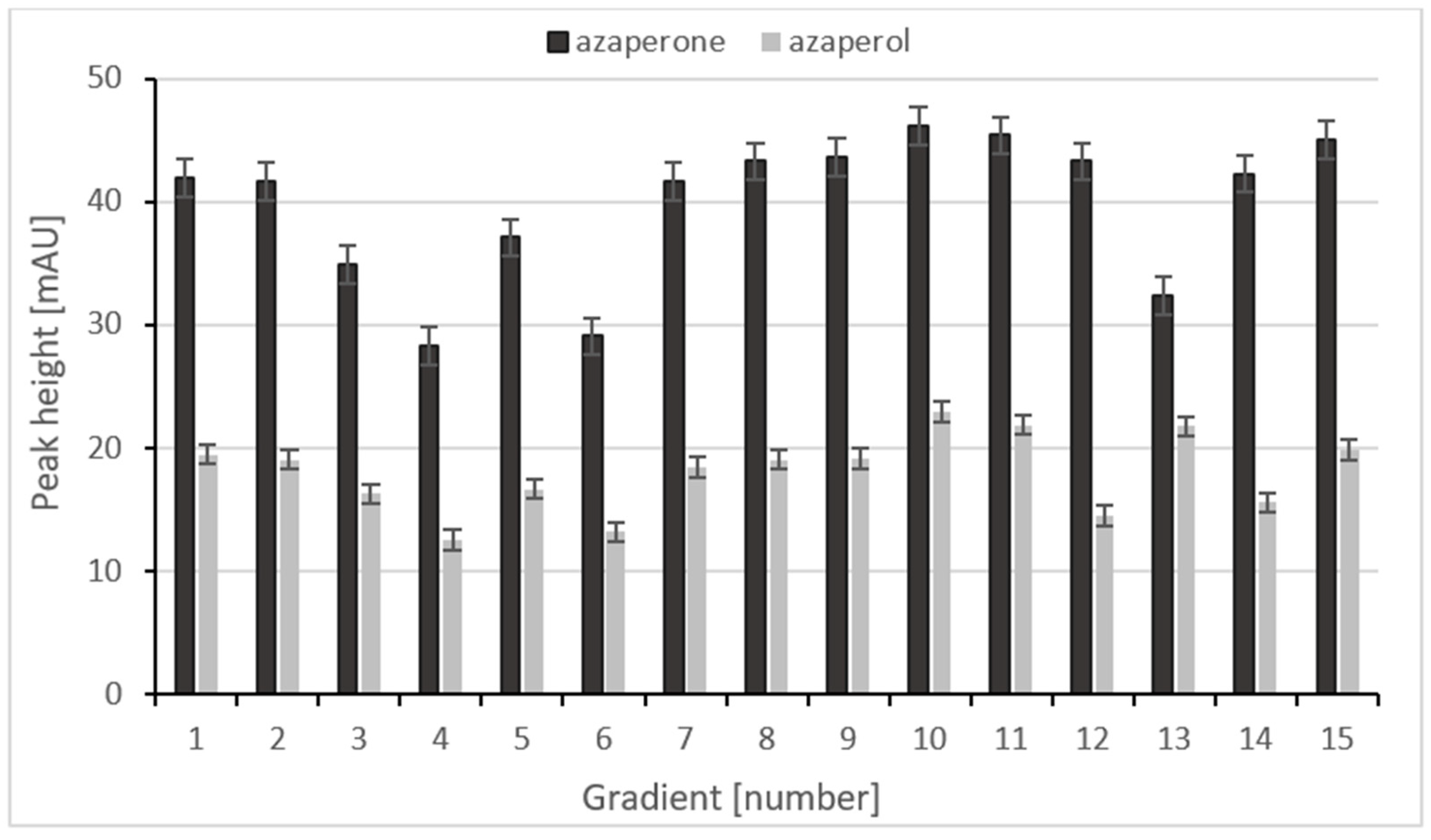
2.2.1. Selection of Mobile Phase
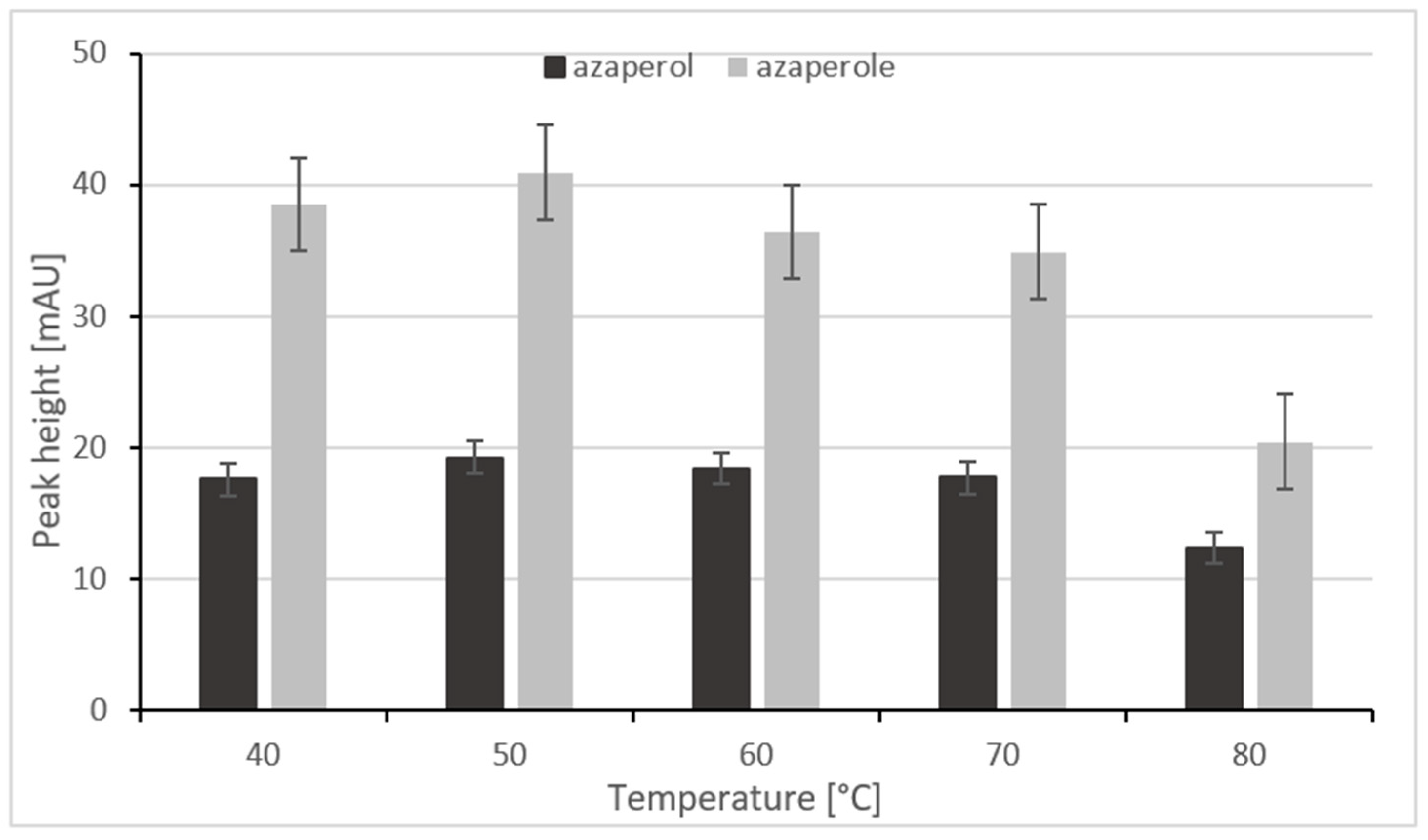
2.2.2. Optimization of Separation Temperature
2.3. Calibration and Other Validation Data
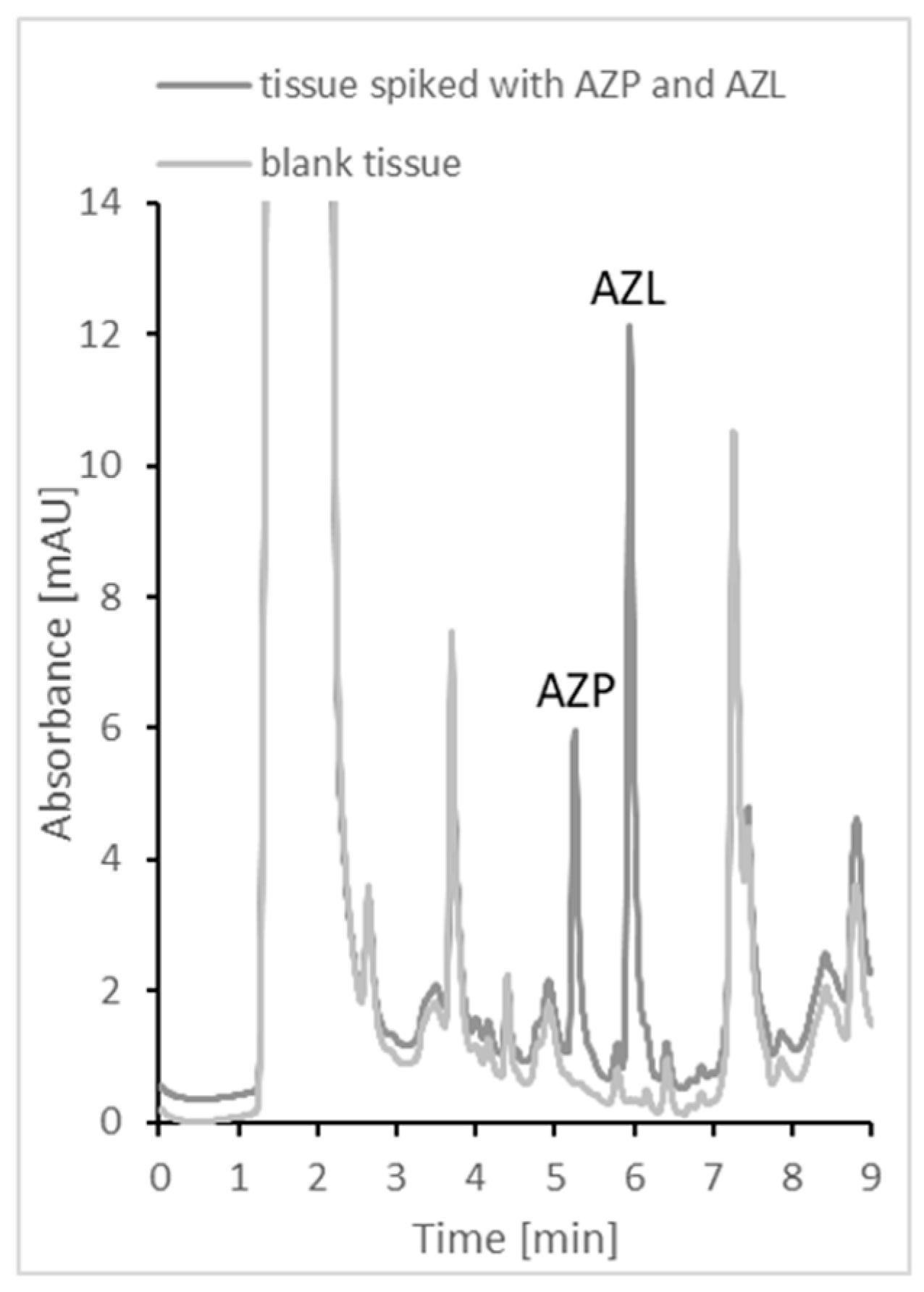
2.4. Application to Real Samples
3. Materials and Methods
3.1. Instruments
3.2. Chemicals
3.3. Chromatographic Conditions
3.4. Sample Collection and Preparation
3.5. Method Validation
3.6. Calibration of the Method
3.7. Greenness
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, L.; Wang, J.; Zhang, G.; Liu, R.; Fang, B. Simultaneous Determination of Tranquilizers and Carazolol Residues in Swine Tissues by Liquid Chromatography-Tandem Mass Spectrometry. Bioanalytical 2012, 45, 1377–1389. [Google Scholar] [CrossRef]
- Brian, K.; O’Mahony, J.; Malone, E.; Moloney, M.; Cantwell, H.; Furey, A.; Danaher, M. Current trends in sample preparation for growth promoter and veterinary drug residue analysis. J. Chromatogr. A 2000, 1216, 7977–8015. [Google Scholar] [CrossRef] [Green Version]
- Scarth, J.P.; Tealea, P.; Kuuranne, T. Drug metabolism in the horse: A review. Drug Test Anal. 2011, 3, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.J.; Perez-Ecija, A.; Toribio, R.E. Clinical Pharmacology in Donkeys and Mules. Vet. Clin. Equine. 2019, 35, 589–606. [Google Scholar] [CrossRef] [PubMed]
- Fluchard, D.; Kiebooms, S.; Dubois, M.; Delahaut, P. Determination of a method for detecting and quantifying azaperone, azaperol and carazolol in pig tissues by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2007, 44, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Sell, B.; Posyniak, A.; Gbylik, M. Screening procedure for simultaneous determination of azaperone, carazolol, and chlorpromazine in animal urine. Bull. Vet. Inst. Pulawy. 2011, 55, 513–517. [Google Scholar]
- Oliveira, L.G.; Barreto, F.; Hoff, R.; Rübensam, G.; Scherer Kurz, M.H.; Galle, G.; Gonçalves, F.F. Validation of a method for sedatives and β-blockers determination in swine, bovine and equine kidney using liquid chromatography coupled with tandem mass spectrometry. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2017, 34, 32–39. [Google Scholar] [CrossRef]
- Cerkvenik-Flajs, V. Determination of residues of azaperone in the kidneys by liquid chromatography with fluorescence detection. Anal. Chim. Acta. 2007, 586, 374–382. [Google Scholar] [CrossRef]
- De Brabander, H.F.; Noppe, H.; Verheyden, K.; Bussche, J.V.; Wille, K.; Okerman, L.; Vanhaecke, L.; Reybroeck, W.; Ooghe, S.; Croubels, S. Residue analysis: Future trends from a historical perspective. J. Chromatogr. A 2009, 1216, 7964–7976. [Google Scholar] [CrossRef]
- Stolker, A.A.M.; Brinkman, U.A.T. Analytical strategies for residue analysis of veterinary drugs and growth-promoting agents in food-producing animals—A review. J. Chromatogr. A 2005, 1067, 15–53. [Google Scholar] [CrossRef]
- Oca, M.L.; Rubio, L.; Ortiz, M.C.; Sarabia, L.A.; García, I.; Bañuelos, M.; Burgos, S. Robustness testing in the determination of seven drugs in animal muscle by liquid chromatography-tandem mass spectrometry. Chemom. Intell. Lab. Syst. 2016, 151, 172–180. [Google Scholar] [CrossRef]
- Bock, C.; Stachel, C.S. Development and validation of a confirmatory method for the determination of tranquilisers and a β-blocker in porcine and bovine kidney by LC-MS/MS Development and validation of a confirmatory method for the determination of tranquilisers and a β-blocker in porcine and bovine kidney by LC-MS/MS. Food Addit. Contam: Part A 2013, 30, 1000–1011. [Google Scholar] [CrossRef]
- Mitrowska, K.; Posyniak, A.; Zmudzki, J. Rapid method for the determination of tranquilizers and a beta-blocker in porcine and bovine kidney by liquid chromatography with tandem mass spectrometry. Anal. Chim. Acta. 2009, 637, 185–192. [Google Scholar] [CrossRef]
- Chen, L.; Hu, X.; Xing, Y.; Sun, Y.; Hu, M.; Zhang, G. Highly sensitive immunochromatographic assay for simultaneous determination of azaperone and azaperol in pork. Food Chem.: X 2023, 17, 100525. [Google Scholar] [CrossRef]
- Keukens, H.J.; Aerts, M.M.L. Determination of residues of carazolol and a number of tranquilizers in swine kidney by high-performance liquid chromatography with ultraviolet and fluorescence detection. J. Chromatogr. A 1991, 464, 149–161. [Google Scholar] [CrossRef]
- Choi, J.H.; Lamshöft, M.; Zühlke, S.; Park, J.H.; Rahman, M.M.; El-Aty, A.M.A.; Spiteller, M.; Shim, J.H. Determination of anxiolytic veterinary drugs from biological fertilizer blood meal using liquid chromatography high-resolution mass spectrometry. Biomed. Chromatogr. 2014, 28, 751–759. [Google Scholar] [CrossRef]
- Choi, J.-H.; Lamshöft, M.; Zühlke, S.; Park, K.H.; Shim, J.-H.; Spiteller, M. Determination of sedatives and adrenergic blockers in blood meal using accelerated solvent extraction and Orbitrap mass spectrometry. J. Chromatogr. A 2012, 1260, 111–119. [Google Scholar] [CrossRef]
- FDA. Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Contains Nonbinding Recommendations; FDA: Washington, DC, USA, 2018. [Google Scholar]
- Delahaut, P.; Levaux, C.; Eloy, P.; Dubois, M. Validation of a method for detecting and quantifying tranquillisers and a-blocker in pig tissues by liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta. 2003, 483, 335–340. [Google Scholar] [CrossRef]
- Delahaut, P.; Brasseur, P.Y.; Dubois, M. Multiresidue method for the detection of tranquillisers, xylazine, and a β-blocker in animal production by liquid chromatography-tandem mass spectrometry. J. Chromatogr. A. 2004, 1054, 373–378. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, L.; Xu, Q.; Wang, S.; Wang, X. Determination of ten sedative residues in pork and kidney by ultra performance liquid chromatography-tandem mass spectrometry. Chinese J. Chromatogr. 2010, 28, 38–42. [Google Scholar] [CrossRef]
- Aokia, Y.; Hakamatab, H.; Igarashia, Y.; Uchidaa, K.; Kobayashia, H.; Hirayamaa, N.; Kotanib, A.; Kusu, F. Simultaneous determination of azaperone and azaperol in animal tissues by HPLC with confirmation by electrospray ionization mass spectrometry. J. Chromatogr. B 2009, 877, 166–172. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Gradient | Gradient Profile |
|---|---|
| 1 | 0–0.5 min, 15% B; 0.5–5 min, 15–50% B; 5–9 min, 50–15% B |
| 2 | 0–0.5 min, 15% B; 0.5–4 min, 15–50% B; 4–9 min, 50–15% B |
| 3 | 0–0.5 min, 15% B; 0.5–5 min, 15–50% B; 5–6 min, 50% B; 6–9 min, 50–15% B |
| 4 | 0–0.5 min, 15% B; 0.5–5 min, 15–30% B; 5–9 min, 30–15% B |
| 5 | 0–0.5 min, 15% B; 0.5–5 min, 15–30% B; 5–7 min, 30–50% B; 7–10 min, 50–15% B |
| 6 | 0–0.5 min, 15% B; 0.5–5 min, 15–30% B; 5–6 min, 30% B; 6–10 min, 30–15% B |
| 7 | 0–0.5 min, 15% B; 0.5–5 min, 15–40% B; 5–10 min, 40–15% B |
| 8 | 0–0.5 min, 15% B; 0.5–5 min, 15–40% B; 5–7 min, 40–50% B; 7–10 min, 50–15% B |
| 9 | 0–0.5 min, 15% B; 0.5–5 min, 15–40% B; 5–6 min, 40% B; 6–10 min, 40–15% B |
| 10 | 0–7 min, 10–50% B; 7–9 min, 50–10% B |
| 11 | 0–1 min, 10–15% B; 1–5 min, 10–15% B; 5–9 min, 50–10% B |
| 12 | 0–0.5 min, 20% B; 0.5–5 min, 20–50% B; 5–9 min, 50–20% B |
| 13 | 0–1 min, 20–30% B; 1–5 min, 30–50% B; 5–9 min, 50–20% B |
| 14 | 0–1 min, 15–20% B; 1–5 min, 20–50% B; 5–9 min, 50–15% B |
| 15 | 0–1 min, 20–30%; 1–5 min, 30–50% B; 5–9 min, 50–12% B |
| Method | LOD (µg/kg) | LOQ (µg/kg) | R2 | Precision [%] | Accuracy [%] | ||
|---|---|---|---|---|---|---|---|
| AZN | AZL | AZN | AZL | ||||
| LC-MS/MS [2] | 0.06–0.1 | 0.1 | 0.2–0.4 | 0.4 | >0.99 | <15 | 74.2–91.8 |
| LC-MS/MS [7] | 0.5 | 0.5 | 2.5 | 2.5 | 0.9826–0.9965 | 1.1–16.6 | 69.8–85.5 |
| LC-FLD [8] | 10 | 3 | 10 | 5 | >0.99 | <11.0 | 88.2–91.2 |
| LC-UV [15] | 1 | no data | no data | no data | 0.997 | 0.6–14.6 | 97.0–112.9 |
| Presented method | 1.0 | 0.4 | 2.5 | 1.2 | 0.9985–0.9991 | 2.6–9.9 | 93.4–109.5 |
| Added * [µg/kg] | Intra-Day | Inter-Day | ||||
|---|---|---|---|---|---|---|
| Found ± SD [µg/kg] | CV [%] | Accuracy [%] | Found ± SD [µg/kg] | CV [%] | Accuracy [%] | |
| Azaperone | ||||||
| 80 | 78.51 ± 2.60 | 3.3 | 98.1 | 81.51 ± 4.50 | 5.5 | 101.9 |
| 150 | 156.59 ± 5.20 | 3.3 | 104.4 | 158.09 ± 4.50 | 2.9 | 105.4 |
| 250 | 273.71 ± 18.20 | 6.7 | 109.5 | 257.19 ± 20.64 | 8.0 | 102.9 |
| Azaperol | ||||||
| 80 | 74.74 ± 4.93 | 6.6 | 93.4 | 75.39 ± 6.30 | 8.4 | 94.2 |
| 150 | 159.05 ± 9.06 | 5.7 | 106.0 | 151.21 ± 11.32 | 7.5 | 100.8 |
| 250 | 272.12 ± 7.07 | 2.6 | 108.9 | 257.74 ± 25.59 | 9.9 | 103.1 |
| Method Described in the Literature [22] | Presented Method | |
|---|---|---|
| Mobile phase | acetonitrile—0.025% aqueous diethylamine mixture (2:3, v/v) | 0.05 mol/L phosphate buffer pH 3 (component A) and acetonitrile (component B) |
| Column/Stationary phase | ODS column (Asahipak ODP-50 4D, 150 mm × 4.6 mm, Showa Denko K.K., Kanagawa, Japan) | Zorbax SB C-18 (150 mm × 4.6 mm, 5 µm, Agilent Technologies). |
| Linearity | 0.05–2 µg/mL | 50–300 µg/kg of tissue |
| Applications | Tissues analysis | Tissues analysis |
| Merits | the method is applicable, good accuracy and precision, confirmation with LC/MS | not time consuming, easy to perform, does not require sophisticated equipment, good accuracy, good precision, lower LOD |
| Demerits | more steps in sample preparation, higher LOD, large volumes of toxic acetonitryle, UV-Vis detection (small concentration sensitivity) | large volumes of toxic acetonitryle, UV-Vis detection (small concentration sensitivity) |
| Sample Number | Added * [µg/kg] | Found ± SD [µg/kg] | CV [%] |
|---|---|---|---|
| Azaperone | |||
| 1 | 230.00 | 218.15 ± 9.38 | 4.3 |
| 2 | 230.00 | 222.65 ± 2.60 | 1.2 |
| 3 | 230.00 | 225.66 ± 4.50 | 2.0 |
| 4 | 230.00 | 214.40 ± 3.19 | 1.5 |
| 5 | 230.00 | 227.16 ± 2.60 | 1.1 |
| 6 | 230.00 | 225.66 ± 7.80 | 3.5 |
| 7 | 230.00 | 236.17 ± 5.20 | 2.2 |
| 8 | 230.00 | 222.65 ± 6.88 | 3.1 |
| 9 | 230.00 | 234.67 ± 13.51 | 5.8 |
| 10 | 230.00 | 212.14 ± 7.80 | 3.7 |
| Azaperol | |||
| 1 | 230.00 | 225.06 ± 7.84 | 3.5 |
| 2 | 230.00 | 225.06 ± 7.07 | 3.1 |
| 3 | 230.00 | 223.10 ± 10.38 | 4.7 |
| 4 | 230.00 | 217.22 ± 8.32 | 3.8 |
| 5 | 230.00 | 223.10 ± 3.92 | 1.8 |
| 6 | 230.00 | 221.80 ± 9.67 | 4.4 |
| 7 | 230.00 | 234.21 ± 6.30 | 2.7 |
| 8 | 230.00 | 217.22 ± 5.55 | 2.6 |
| 9 | 230.00 | 234.21 ± 6.30 | 2.7 |
| 10 | 230.00 | 213.30 ± 3.92 | 1.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kośka, I.; Kubalczyk, P. Development of the Chromatographic Method for Simultaneous Determination of Azaperone and Azaperol in Animal Kidneys and Livers. Int. J. Mol. Sci. 2023, 24, 100. https://doi.org/10.3390/ijms24010100
Kośka I, Kubalczyk P. Development of the Chromatographic Method for Simultaneous Determination of Azaperone and Azaperol in Animal Kidneys and Livers. International Journal of Molecular Sciences. 2023; 24(1):100. https://doi.org/10.3390/ijms24010100
Chicago/Turabian StyleKośka, Izabella, and Paweł Kubalczyk. 2023. "Development of the Chromatographic Method for Simultaneous Determination of Azaperone and Azaperol in Animal Kidneys and Livers" International Journal of Molecular Sciences 24, no. 1: 100. https://doi.org/10.3390/ijms24010100