
Non-Traditional Pathways for Platelet Pathophysiology in Diabetes: Implications for Future Therapeutic Targets


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathophysiology of Vascular Disease in Diabetes
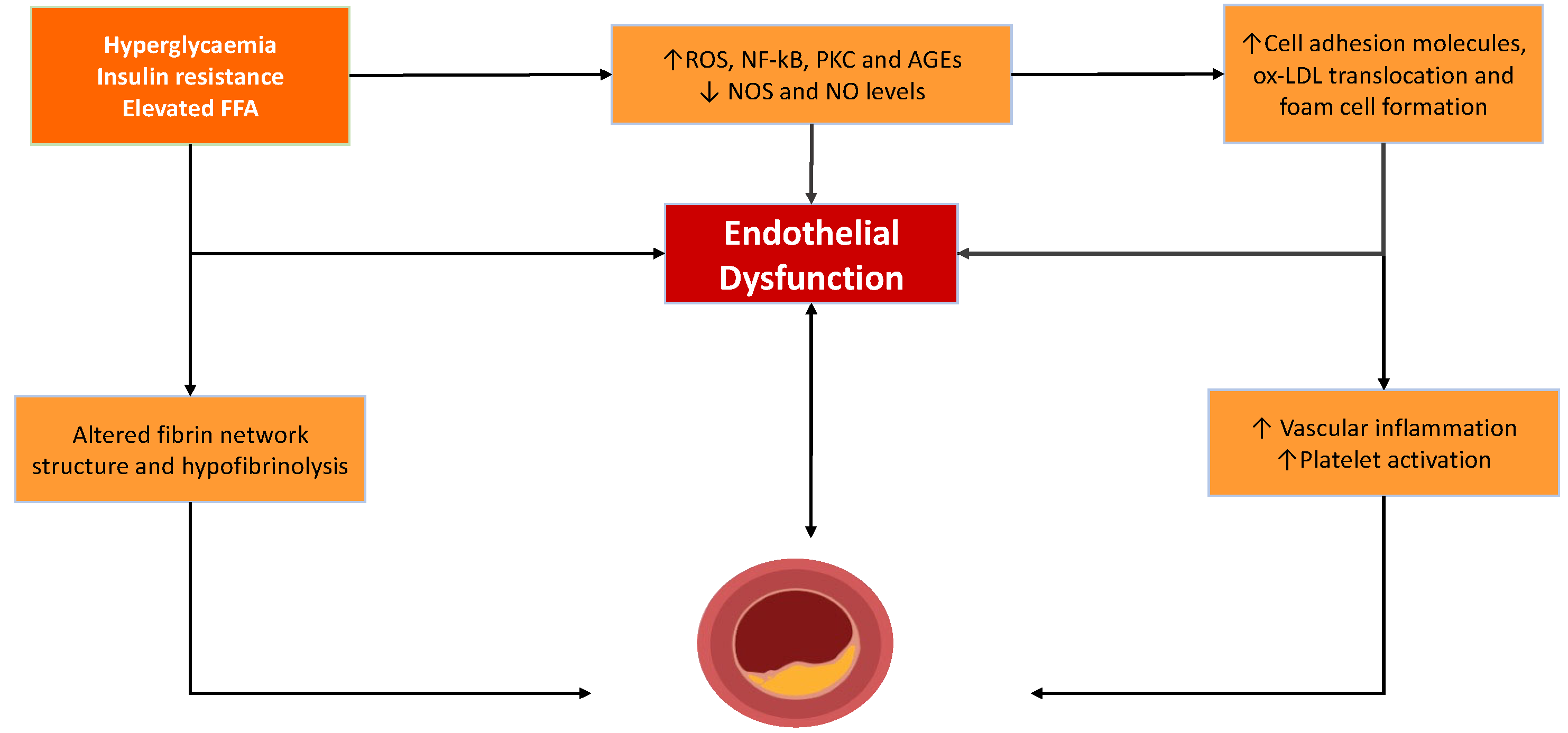
2.1. Endothelial Dysfunction and Atheroma Formation
2.2. Intravascular Thrombus Formation
3. Diabetes-Related Mechanistic Pathways Modulating Thrombo-Inflammatory Function of Platelet
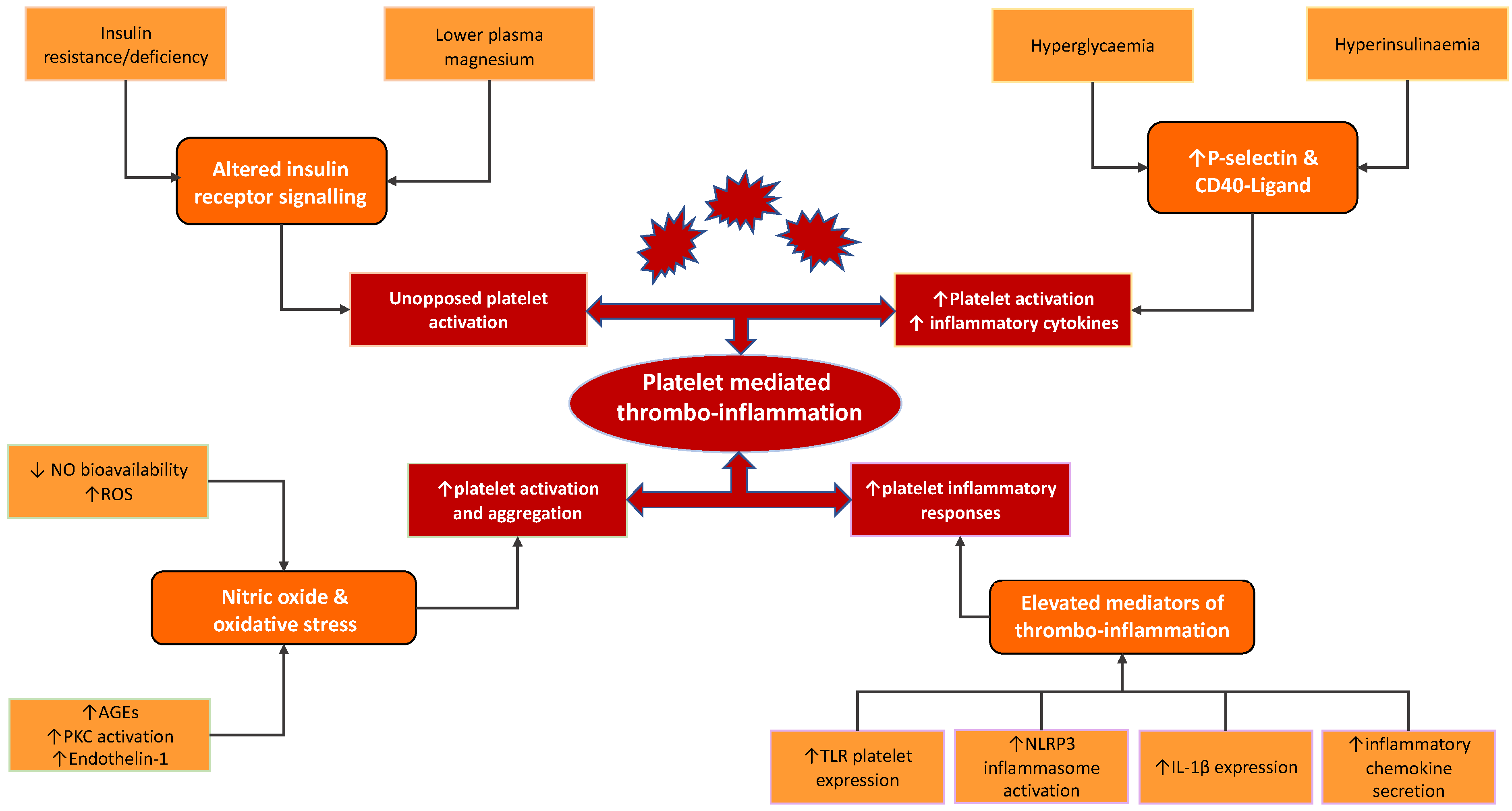
3.1. Insulin and the Insulin Receptor
3.2. Nitric Oxide and Reactive Oxygen Species
3.3. Platelet Activation and P-Selectin
3.4. CD40-Ligand
3.5. Toll-like Receptors and Immune Response
4. Platelet Metabolic Reprogramming in Diabetes: Future Therapeutic Targets?
4.1. Metabolic Reprogramming and Platelet Bioenergetics
4.2. Altered Platelet mRNA and Protein Expression
4.3. Platelet-Specific miRNA
4.4. Mitochondrial Dysfunction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bulugahapitiya, U.; Siyambalapitiya, S.; Sithole, J.; Idris, I. Is diabetes a coronary risk equivalent? Systematic review and meta-analysis. Diabet. Med. 2009, 26, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Pajunen, P.; Koukkunen, H.; Ketonen, M.; Jerkkola, T.; Immonen-Raiha, P.; Karja-Koskenkari, P.; Kuulasmaa, K.; Palomaki, P.; Mustonen, J.; Lehtonen, A.; et al. Myocardial infarction in diabetic and non-diabetic persons with and without prior myocardial infarction: The FINAMI Study. Diabetologia 2005, 48, 2519–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hex, N.; Bartlett, C.; Wright, D.; Taylor, M.; Varley, D. Estimating the current and future costs of Type 1 and Type 2 diabetes in the UK, including direct health costs and indirect societal and productivity costs. Diabet. Med. 2012, 29, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Cubbon, R.M.; Wheatcroft, S.B.; Grant, P.J.; Gale, C.P.; Barth, J.H.; Sapsford, R.J.; Ajjan, R.; Kearney, M.T.; Hall, A.S. On behalf of EMMACE Investigators. Temporal trends in mortality of patients with diabetes mellitus suffering acute myocardial infarction: A comparison of over 3000 patients between 1995 and 2003. Eur. Heart J. 2007, 28, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J. Factors contributing to increased platelet reactivity in people with diabetes. Diabetes Care 2009, 32, 525–527. [Google Scholar] [CrossRef] [Green Version]
- Angiolillo, D.J.; Roffi, M.; Fernandez-Ortiz, A. Tackling the thrombotic burden in patients with acute coronary syndrome and diabetes mellitus. Expert Rev. Cardiovasc. 2011, 9, 697–710. [Google Scholar] [CrossRef]
- Franchi, F.; Angiolillo, D.J. Novel antiplatelet agents in acute coronary syndrome. Nat. Rev. Cardiol. 2015, 12, 30–47. [Google Scholar] [CrossRef]
- Angiolillo, D.J.; Ueno, M.; Goto, S. Basic principles of platelet biology and clinical implications. Circ. J. 2010, 74, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Capodanno, D.; Angiolillo, D.J. Aspirin for Primary Cardiovascular Risk Prevention and Beyond in Diabetes Mellitus. Circulation 2016, 134, 1579–1594. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar] [CrossRef]
- Margraf, A.; Zarbock, A. Platelets in Inflammation and Resolution. J. Immunol. 2019, 203, 2357–2367. [Google Scholar] [CrossRef]
- Patko, Z.; Csaszar, A.; Acsady, G.; Ory, I.; Takacs, E.; Furesz, J. Elevation of monocyte-platelet aggregates is an early marker of type 2 diabetes. Interv. Med. Appl. Sci. 2012, 4, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Zahran, A.M.; El-Badawy, O.; Mohamad, I.L.; Tamer, D.M.; Abdel-Aziz, S.M.; Elsayh, K.I. Platelet Activation and Platelet-Leukocyte Aggregates in Type I Diabetes Mellitus. Clin. Appl. Thromb. Hemost. 2018, 24, 230S–239S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondina, M.T.; Weyrich, A.S.; Zimmerman, G.A. Platelets as Cellular Effectors of Inflammation in Vascular Diseases. Circ. Res. 2013, 112, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Creager, M.A.; Lüscher, T.F.; Cosentino, F.; Beckman, J.A. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Circulation 2003, 108, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Rondina, M.T. The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells during Infectious Diseases. Front. Immunol. 2019, 10, 2204. [Google Scholar] [CrossRef] [Green Version]
- Pechlivani, N.; Ajjan, R.A. Thrombosis and Vascular Inflammation in Diabetes: Mechanisms and Potential Therapeutic Targets. Front. Cardiovasc. Med. 2018, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Carrizzo, A.; Izzo, C.; Oliveti, M.; Alfano, A.; Virtuoso, N.; Capunzo, M.; Di Pietro, P.; Calabrese, M.; De Simone, E.; Sciarretta, S.; et al. The Main Determinants of Diabetes Mellitus Vascular Complications: Endothelial Dysfunction and Platelet Hyperaggregation. Int. J. Mol. Sci. 2018, 19, 2968. [Google Scholar] [CrossRef] [Green Version]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef]
- Hartge, M.M.; Unger, T.; Kintscher, U. The endothelium and vascular inflammation in diabetes. Diabete. Vasc. Dis. Res. 2007, 4, 84–88. [Google Scholar] [CrossRef]
- Hadi, H.A.; Suwaidi, J.A. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 2007, 3, 853–876. [Google Scholar] [PubMed]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef] [PubMed]
- Colwell, J.A.; Nesto, R.W. The platelet in diabetes: Focus on prevention of ischemic events. Diabetes Care 2003, 26, 2181–2188. [Google Scholar] [CrossRef] [Green Version]
- King, R.J.; Grant, P.J. Diabetes and cardiovascular disease: Pathophysiology of a life-threatening epidemic. Herz 2016, 41, 184–192. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T. Inflammation and coagulation. Crit. Care Med. 2010, 38, S26–S34. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Hess, K.; Grant, P.J. Inflammation and thrombosis in diabetes. Thromb. Haemost. 2011, 105 (Suppl. S1), S43–S54. [Google Scholar] [CrossRef]
- Kearney, K.; Tomlinson, D.; Smith, K.; Ajjan, R. Hypofibrinolysis in diabetes: A therapeutic target for the reduction of cardiovascular risk. Cardiovasc. Diabetol. 2017, 16, 34. [Google Scholar] [CrossRef] [Green Version]
- King, R.; Ajjan, R. Hypoglycaemia, thrombosis and vascular events in diabetes. Expert Rev. Cardiovasc. Ther. 2016, 14, 1099–1101. [Google Scholar] [CrossRef] [Green Version]
- Kurdee, Z.; King, R.; Ajjan, R.A. The fibrin network in diabetes: Its role in thrombosis risk. Pol. Arch. Med. Wewn. 2014, 124, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.H.; Hess, K.; Price, J.F.; Strachan, M.; Baxter, P.D.; Cubbon, R.; Phoenix, F.; Gamlen, T.; Ariens, R.A.; Grant, P.J.; et al. Gender-specific alterations in fibrin structure function in type 2 diabetes: Associations with cardiometabolic and vascular markers. J. Clin. Endocrinol. Metab. 2012, 97, E2282–E2287. [Google Scholar] [CrossRef] [PubMed]
- Sumaya, W.; Wallentin, L.; James, S.K.; Siegbahn, A.; Gabrysch, K.; Himmelmann, A.; Ajjan, R.A.; Storey, R.F. Impaired Fibrinolysis Predicts Adverse Outcome in Acute Coronary Syndrome Patients with Diabetes: A PLATO Sub-Study. Thromb. Haemost. 2020, 120, 412–422. [Google Scholar] [CrossRef] [Green Version]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Tabák, A.G.; Jokela, M.; Akbaraly, T.N.; Brunner, E.J.; Kivimäki, M.; Witte, D.R. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: An analysis from the Whitehall II study. Lancet 2009, 373, 2215–2221. [Google Scholar] [CrossRef] [Green Version]
- Westerbacka, J.; Yki-Jarvinen, H.; Turpeinen, A.; Rissanen, A.; Vehkavaara, S.; Syrjala, M.; Lassila, R. Inhibition of platelet-collagen interaction: An in vivo action of insulin abolished by insulin resistance in obesity. Arter. Thromb. Vasc. Biol. 2002, 22, 167–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisinski, J.A.; Kimple, M.E. Platelet Dysfunction in Type 1 Diabetes: Stressing the Thromboxanes. Diabetes 2016, 65, 349–351. [Google Scholar] [CrossRef] [Green Version]
- Alessandrini, P.; McRae, J.; Feman, S.; FitzGerald, G.A. Thromboxane biosynthesis and platelet function in type I diabetes mellitus. N. Engl. J. Med. 1988, 319, 208–212. [Google Scholar] [CrossRef]
- Hu, H.; Li, N.; Ekberg, K.; Johansson, B.L.; Hjemdahl, P. Insulin, but not proinsulin C-peptide, enhances platelet fibrinogen binding in vitro in Type 1 diabetes mellitus patients and healthy subjects. Thromb. Res. 2002, 106, 91–95. [Google Scholar] [CrossRef]
- Vignini, A.; Moroni, C.; Nanetti, L.; Raffaelli, F.; Cester, A.; Gabrielli, O.; Cherubini, V.; Mazzanti, L. Alterations of platelet biochemical and functional properties in newly diagnosed type 1 diabetes: A role in cardiovascular risk? Diabetes Metab. Res. Rev. 2011, 27, 277–285. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Phoenix, F.A.; Pitt, S.J.; Ajjan, R.A.; Stewart, A.J. Reduced Plasma Magnesium Levels in Type-1 Diabetes Associate with Prothrombotic Changes in Fibrin Clotting and Fibrinolysis. Thromb. Haemost. 2020, 120, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, I.A.; Mocking, A.I.M.; Feijge, M.A.H.; Gorter, G.; Haeften, T.W.v.; Heemskerk, J.W.M.; Akkerman, J.-W.N. Platelet Inhibition by Insulin Is Absent in Type 2 Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapp, B.R.; Hingorani, A.D.; Kharbanda, R.K.; Mohamed-Ali, V.; Stephens, J.W.; Vallance, P.; MacAllister, R.J. Inflammation-induced endothelial dysfunction involves reduced nitric oxide bioavailability and increased oxidant stress. Cardiovasc. Res. 2004, 64, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Agbani, E.O.; Coats, P.; Mills, A.; Wadsworth, R.M. Peroxynitrite stimulates pulmonary artery endothelial and smooth muscle cell proliferation: Involvement of ERK and PKC. Pulm. Pharmacol. 2011, 24, 100–109. [Google Scholar] [CrossRef]
- Pacher, P.; Szabó, C. Role of peroxynitrite in the pathogenesis of cardiovascular complications of diabetes. Curr. Opin. Pharm. 2006, 6, 136–141. [Google Scholar] [CrossRef]
- Agbani, E.O.; Coats, P.; Wadsworth, R.M. Acute hypoxia stimulates intracellular peroxynitrite formation associated with pulmonary artery smooth muscle cell proliferation. J. Cardiovasc. Pharm. 2011, 57, 584–588. [Google Scholar] [CrossRef]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef]
- Zeiher, A.M.; Fisslthaler, B.; Schray-Utz, B.; Busse, R. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ. Res. 1995, 76, 980–986. [Google Scholar] [CrossRef]
- Schafer, A.; Alp, N.J.; Cai, S.; Lygate, C.A.; Neubauer, S.; Eigenthaler, M.; Bauersachs, J.; Channon, K.M. Reduced vascular NO bioavailability in diabetes increases platelet activation in vivo. Arter. Thromb. Vasc. Biol. 2004, 24, 1720–1726. [Google Scholar] [CrossRef] [Green Version]
- Ayub, T.; Khan, S.N.; Ayub, S.G.; Dar, R.; Andrabi, K.I. Reduced nitrate level in individuals with hypertension and diabetes. J. Cardiovasc. Dis. Res. 2011, 2, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Rodrigues-Krause, J.; O’Hagan, C.; De Vito, G.; Boreham, C.; Susta, D.; Newsholme, P.; Murphy, C. Differential nitric oxide levels in the blood and skeletal muscle of type 2 diabetic subjects may be consequence of adiposity: A preliminary study. Metabolism 2012, 61, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Assert, R.; Scherk, G.; Bumbure, A.; Pirags, V.; Schatz, H.; Pfeiffer, A.F. Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia 2001, 44, 188–195. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davì, G.; Catalano, I.; Averna, M.; Notarbartolo, A.; Strano, A.; Ciabattoni, G.; Patrono, C. Thromboxane Biosynthesis and Platelet Function in Type II Diabetes Mellitus. N. Engl. J. Med. 1990, 322, 1769–1774. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Rini, G.B.; Averna, M.; Novo, S.; Di Fede, G.; Pinto, A.; Notarbartolo, A.; Strano, A. Thromboxane B2 formation and platelet sensitivity to prostacyclin in insulin-dependent and insulin-independent diabetics. Thromb. Res. 1982, 26, 359–370. [Google Scholar] [CrossRef]
- Davì, G.; Gresele, P.; Violi, F.; Basili, S.; Catalano, M.; Giammarresi, C.; Volpato, R.; Nenci, G.G.; Ciabattoni, G.; Patrono, C. Diabetes Mellitus, Hypercholesterolemia, and Hypertension but Not Vascular Disease Per Se Are Associated with Persistent Platelet Activation In Vivo. Circulation 1997, 96, 69–75. [Google Scholar] [CrossRef]
- Santilli, F.; Formoso, G.; Sbraccia, P.; Averna, M.; Miccoli, R.; Di Fulvio, P.; Ganci, A.; Pulizzi, N.; Lattanzio, S.; Ciabattoni, G.; et al. Postprandial hyperglycemia is a determinant of platelet activation in early type 2 diabetes mellitus. J. Thromb. Haemost. 2010, 8, 828–837. [Google Scholar] [CrossRef]
- Vieira-de-Abreu, A.; Campbell, R.A.; Weyrich, A.S.; Zimmerman, G.A. Platelets: Versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin. Immunopathol. 2012, 34, 5–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyula, V.R.; Boden, G.; Rao, A.K. Platelet and monocyte activation by hyperglycemia and hyperinsulinemia in healthy subjects. Platelets 2006, 17, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Yngen, M.; Norhammar, A.; Hjemdahl, P.; Wallen, N.H. Effects of improved metabolic control on platelet reactivity in patients with type 2 diabetes mellitus following coronary angioplasty. Diab. Vasc. Dis. Res. 2006, 3, 52–56. [Google Scholar] [CrossRef]
- Eibl, N.; Krugluger, W.; Streit, G.; Schrattbauer, K.; Hopmeier, P.; Schernthaner, G. Improved metabolic control decreases platelet activation markers in patients with type-2 diabetes. Eur. J. Clin. Investig. 2004, 34, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, L.; Thomson, G.J.A.; Adams, R.C.M.; Nell, T.A.; Laubscher, W.A.; Pretorius, E. Platelet activity and hypercoagulation in type 2 diabetes. Cardiovasc. Diabetol. 2018, 17, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloui, C.; Prigent, A.; Sut, C.; Tariket, S.; Hamzeh-Cognasse, H.; Pozzetto, B.; Richard, Y.; Cognasse, F.; Laradi, S.; Garraud, O. The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int. J. Mol. Sci. 2014, 15, 22342–22364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiener, P.A.; Moran-Davis, P.; Rankin, B.M.; Wahl, A.F.; Aruffo, A.; Hollenbaugh, D. Stimulation of CD40 with purified soluble gp39 induces proinflammatory responses in human monocytes. J. Immunol. 1995, 155, 4917–4925. [Google Scholar]
- Harding, S.A.; Sommerfield, A.J.; Sarma, J.; Twomey, P.J.; Newby, D.E.; Frier, B.M.; Fox, K.A. Increased CD40 ligand and platelet-monocyte aggregates in patients with type 1 diabetes mellitus. Atherosclerosis 2004, 176, 321–325. [Google Scholar] [CrossRef]
- Varo, N.; Vicent, D.; Libby, P.; Nuzzo, R.; Calle-Pascual, A.L.; Bernal, M.R.; Fernández-Cruz, A.; Veves, A.; Jarolim, P.; Varo, J.J.; et al. Elevated plasma levels of the atherogenic mediator soluble CD40 ligand in diabetic patients: A novel target of thiazolidinediones. Circulation 2003, 107, 2664–2669. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Horigome, H.; Tanaka, K.; Nakata, Y.; Ohkawara, K.; Katayama, Y.; Matsui, A. Impact of weight reduction on production of platelet-derived microparticles and fibrinolytic parameters in obesity. Thromb. Res. 2007, 119, 45–53. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davi, G.; Guagnano, M.T.; Ciabattoni, G.; Basili, S.; Falco, A.; Marinopiccoli, M.; Nutini, M.; Sensi, S.; Patrono, C. Platelet activation in obese women: Role of inflammation and oxidant stress. JAMA 2002, 288, 2008–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santilli, F.; Davi, G.; Consoli, A.; Cipollone, F.; Mezzetti, A.; Falco, A.; Taraborelli, T.; Devangelio, E.; Ciabattoni, G.; Basili, S.; et al. Thromboxane-dependent CD40 ligand release in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2006, 47, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heger, L.A.; Hortmann, M.; Albrecht, M.; Colberg, C.; Peter, K.; Witsch, T.; Stallmann, D.; Zirlik, A.; Bode, C.; Duerschmied, D.; et al. Inflammation in acute coronary syndrome: Expression of TLR2 mRNA is increased in platelets of patients with ACS. PLoS ONE 2019, 14, e0224181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurses, K.M.; Kocyigit, D.; Yalcin, M.U.; Canpinar, H.; Oto, M.A.; Ozer, N.; Tokgozoglu, L.; Guc, D.; Aytemir, K. Enhanced Platelet Toll-like Receptor 2 and 4 Expression in Acute Coronary Syndrome and Stable Angina Pectoris. Am. J. Cardiol. 2015, 116, 1666–1671. [Google Scholar] [CrossRef]
- Rivadeneyra, L.; Carestia, A.; Etulain, J.; Pozner, R.G.; Fondevila, C.; Negrotto, S.; Schattner, M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb. Res. 2014, 133, 235–243. [Google Scholar] [CrossRef]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef]
- Herder, C.; Dalmas, E.; Boni-Schnetzler, M.; Donath, M.Y. The IL-1 Pathway in Type 2 Diabetes and Cardiovascular Complications. Trends Endocrinol. Metab. 2015, 26, 551–563. [Google Scholar] [CrossRef]
- Camell, C.; Goldberg, E.; Dixit, V.D. Regulation of Nlrp3 inflammasome by dietary metabolites. Semin. Immunol. 2015, 27, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Gora, I.M.; Ciechanowska, A.; Ladyzynski, P. NLRP3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes. Cells 2021, 10, 314. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes Metab. Syndr. Obes. 2019, 12, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bester, J.; Pretorius, E. Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci. Rep. 2016, 6, 32188. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Le, Y.; Chen, H.; Zhu, J.; Lu, D. Role of PKM2-Mediated Immunometabolic Reprogramming on Development of Cytokine Storm. Front. Immunol. 2021, 12, 748573. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; O’neill, L.A. The Warburg effect then and now: From cancer to inflammatory diseases. Bioessays 2013, 35, 965–973. [Google Scholar] [CrossRef]
- Lam, F.W.; Vijayan, K.V.; Rumbaut, R.E. Platelets and Their Interactions with Other Immune Cells. Compr. Physiol. 2015, 5, 1265–1280. [Google Scholar] [CrossRef] [Green Version]
- Duerschmied, D.; Bode, C.; Ahrens, I. Immune functions of platelets. Thromb. Haemost. 2014, 112, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Aibibula, M.; Naseem, K.M.; Sturmey, R.G. Glucose metabolism and metabolic flexibility in blood platelets. J. Thromb. Haemost. 2018, 16, 2300–2314. [Google Scholar] [CrossRef] [Green Version]
- Fidler, T.P.; Marti, A.; Gerth, K.; Middleton, E.A.; Campbell, R.A.; Rondina, M.T.; Weyrich, A.S.; Abel, E.D. Glucose Metabolism Is Required for Platelet Hyperactivation in a Murine Model of Type 1 Diabetes. Diabetes 2019, 68, 932–938. [Google Scholar] [CrossRef]
- Guo, X.; Wu, J.; Du, J.; Ran, J.; Xu, J. Platelets of type 2 diabetic patients are characterized by high ATP content and low mitochondrial membrane potential. Platelets 2009, 20, 588–593. [Google Scholar] [CrossRef]
- Chacko, B.K.; Smith, M.R.; Johnson, M.S.; Benavides, G.; Culp, M.L.; Pilli, J.; Shiva, S.; Uppal, K.; Go, Y.-M.; Jones, D.P.; et al. Mitochondria in precision medicine; linking bioenergetics and metabolomics in platelets. Redox Biol. 2019, 22, 101165. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [Green Version]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101–e111. [Google Scholar] [CrossRef] [Green Version]
- Raghavachari, N.; Xu, X.; Harris, A.; Villagra, J.; Logun, C.; Barb, J.; Solomon, M.A.; Suffredini, A.F.; Danner, R.L.; Kato, G.; et al. Amplified expression profiling of platelet transcriptome reveals changes in arginine metabolic pathways in patients with sickle cell disease. Circulation 2007, 115, 1551–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lood, C.; Amisten, S.; Gullstrand, B.; Jönsen, A.; Allhorn, M.; Truedsson, L.; Sturfelt, G.; Erlinge, D.; Bengtsson, A.A. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: Up-regulation of the type I interferon system is strongly associated with vascular disease. Blood 2010, 116, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Marcantoni, E.; Allen, N.; Cambria, M.R.; Dann, R.; Cammer, M.; Lhakhang, T.; O’Brien, M.P.; Kim, B.; Worgall, T.; Heguy, A.; et al. Platelet Transcriptome Profiling in HIV and ATP-Binding Cassette Subfamily C Member 4 (ABCC4) as a Mediator of Platelet Activity. JACC Basic Transl. Sci. 2018, 3, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Mirhashemi, M.E.; Shah, R.V.; Kitchen, R.R.; Rong, J.; Spahillari, A.; Pico, A.R.; Vitseva, O.; Levy, D.; Demarco, D.; Shah, S.; et al. The Dynamic Platelet Transcriptome in Obesity and Weight Loss. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 854–864. [Google Scholar] [CrossRef]
- Heffron, S.P.; Marier, C.; Parikh, M.; Fisher, E.A.; Berger, J.S. Severe obesity and bariatric surgery alter the platelet mRNA profile. Platelets 2019, 30, 967–974. [Google Scholar] [CrossRef]
- Hosono, M.; de Boer, O.J.; van der Wal, A.C.; van der Loos, C.M.; Teeling, P.; Piek, J.J.; Ueda, M.; Becker, A.E. Increased expression of T cell activation markers (CD25, CD26, CD40L and CD69) in atherectomy specimens of patients with unstable angina and acute myocardial infarction. Atherosclerosis 2003, 168, 73–80. [Google Scholar] [CrossRef]
- Shi, R.; Zhou, X.; Ji, W.J.; Zhang, Y.Y.; Ma, Y.Q.; Zhang, J.Q.; Li, Y.M. The Emerging Role of miR-223 in Platelet Reactivity: Implications in Antiplatelet Therapy. Biomed. Res. Int 2015, 2015, 981841. [Google Scholar] [CrossRef] [PubMed]
- Pordzik, J.; Jakubik, D.; Jarosz-Popek, J.; Wicik, Z.; Eyileten, C.; De Rosa, S.; Indolfi, C.; Siller-Matula, J.M.; Czajka, P.; Postula, M. Significance of circulating microRNAs in diabetes mellitus type 2 and platelet reactivity: Bioinformatic analysis and review. Cardiovasc. Diabetol. 2019, 18, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fejes, Z.; Póliska, S.; Czimmerer, Z.; Káplár, M.; Penyige, A.; Gál Szabó, G.; Beke Debreceni, I.; Kunapuli, S.P.; Kappelmayer, J.; Nagy, B., Jr. Hyperglycaemia suppresses microRNA expression in platelets to increase P2RY12 and SELP levels in type 2 diabetes mellitus. Thromb. Haemost. 2017, 117, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Moura, J.; Børsheim, E.; Carvalho, E. The Role of MicroRNAs in Diabetic Complications—Special Emphasis on Wound Healing. Genes 2014, 5, 926–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgheznawy, A.; Shi, L.; Hu, J.; Wittig, I.; Laban, H.; Pircher, J.; Mann, A.; Provost, P.; Randriamboavonjy, V.; Fleming, I. Dicer Cleavage by Calpain Determines Platelet microRNA Levels and Function in Diabetes. Circ. Res. 2015, 117, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.A.E.; Schulte, C.; Barwari, T.; Phoenix, F.; Pearson, S.M.; Mayr, M.; Grant, P.J.; Storey, R.F.; Ajjan, R.A. Aspirin, clopidogrel and prasugrel monotherapy in patients with type 2 diabetes mellitus: A double-blind randomised controlled trial of the effects on thrombotic markers and microRNA levels. Cardiovasc. Diabetol. 2020, 19, 3. [Google Scholar] [CrossRef]
- Parihar, P.; Shetty, R.; Ghafourifar, P.; Parihar, M.S. Increase in oxidative stress and mitochondrial impairment in hypothalamus of streptozotocin treated diabetic rat: Antioxidative effect of Withania somnifera. Cell. Mol. Biol. 2016, 62, 73–83. [Google Scholar]
- Lee, S.H.; Du, J.; Stitham, J.; Atteya, G.; Lee, S.; Xiang, Y.; Wang, D.; Jin, Y.; Leslie, K.L.; Spollett, G.; et al. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. EMBO Mol. Med. 2016, 8, 779–795. [Google Scholar] [CrossRef]
- Ding, W.X.; Li, M.; Biazik, J.M.; Morgan, D.G.; Guo, F.; Ni, H.M.; Goheen, M.; Eskelinen, E.L.; Yin, X.M. Electron microscopic analysis of a spherical mitochondrial structure. J. Biol. Chem. 2012, 287, 42373–42378. [Google Scholar] [CrossRef] [Green Version]
- Avila, C.; Huang, R.J.; Stevens, M.V.; Aponte, A.M.; Tripodi, D.; Kim, K.Y.; Sack, M.N. Platelet mitochondrial dysfunction is evident in type 2 diabetes in association with modifications of mitochondrial anti-oxidant stress proteins. Exp. Clin. Endocrinol. Diabetes 2012, 120, 248–251. [Google Scholar] [CrossRef]
- Bynum, J.A.; Meledeo, M.A.; Getz, T.M.; Rodriguez, A.C.; Aden, J.K.; Cap, A.P.; Pidcoke, H.F. Bioenergetic profiling of platelet mitochondria during storage: 4°C storage extends platelet mitochondrial function and viability. Transfusion 2016, 56 (Suppl. S1), S76–S84. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; Atteya, G.; Gleim, S.; Spollett, G.; Martin, K.; et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation 2014, 129, 1598–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front. Pharm. 2012, 3, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Stitham, J.; Gleim, S.; Di Febbo, C.; Porreca, E.; Fava, C.; Tacconelli, S.; Capone, M.; Evangelista, V.; Levantesi, G.; et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Investig. 2011, 121, 4462–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aune, D.; Keum, N.; Giovannucci, E.; Fadnes, L.T.; Boffetta, P.; Greenwood, D.C.; Tonstad, S.; Vatten, L.J.; Riboli, E.; Norat, T. Dietary intake and blood concentrations of antioxidants and the risk of cardiovascular disease, total cancer, and all-cause mortality: A systematic review and dose-response meta-analysis of prospective studies. Am. J. Clin. Nutr. 2018, 108, 1069–1091. [Google Scholar] [CrossRef]
- Bohn, T. Carotenoids and Markers of Oxidative Stress in Human Observational Studies and Intervention Trials: Implications for Chronic Diseases. Antioxidants 2019, 8, 179. [Google Scholar] [CrossRef] [Green Version]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.E.; Gerstein, H.C.; et al. Effects of Vitamin E on Cardiovascular and Microvascular Outcomes in High-Risk Patients with Diabetes: Results of the HOPE Study and MICRO-HOPE Substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, L.M.; Lin, E.; Mick, E.; Koupenova, M.; Weinberg, E.O.; Kramer, C.D.; Genco, C.A.; Tanriverdi, K.; Larson, M.G.; Benjamin, E.J.; et al. Interleukin 1 receptor 1 and interleukin 1beta regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Ajjan, R.; Storey, R.F.; Grant, P.J. Aspirin resistance and diabetes mellitus. Diabetologia 2008, 51, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Ertugrul, D.T.; Tutal, E.; Yıldız, M.; Akin, O.; Yalçın, A.A.; Üre, O.Z.S.; Yılmaz, H.; Yavuz, B.N.; Deveci, O.S.; Ata, N.; et al. Aspirin Resistance Is Associated with Glycemic Control, the Dose of Aspirin, and Obesity in Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2010, 95, 2897–2901. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagar, R.C.; Ajjan, R.A.; Naseem, K.M. Non-Traditional Pathways for Platelet Pathophysiology in Diabetes: Implications for Future Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 4973. https://doi.org/10.3390/ijms23094973
Sagar RC, Ajjan RA, Naseem KM. Non-Traditional Pathways for Platelet Pathophysiology in Diabetes: Implications for Future Therapeutic Targets. International Journal of Molecular Sciences. 2022; 23(9):4973. https://doi.org/10.3390/ijms23094973
Chicago/Turabian StyleSagar, Rebecca C., Ramzi A. Ajjan, and Khalid M. Naseem. 2022. "Non-Traditional Pathways for Platelet Pathophysiology in Diabetes: Implications for Future Therapeutic Targets" International Journal of Molecular Sciences 23, no. 9: 4973. https://doi.org/10.3390/ijms23094973