
Phosphoinositide 3-Kinases as Potential Targets for Thrombosis Prevention


Abstract
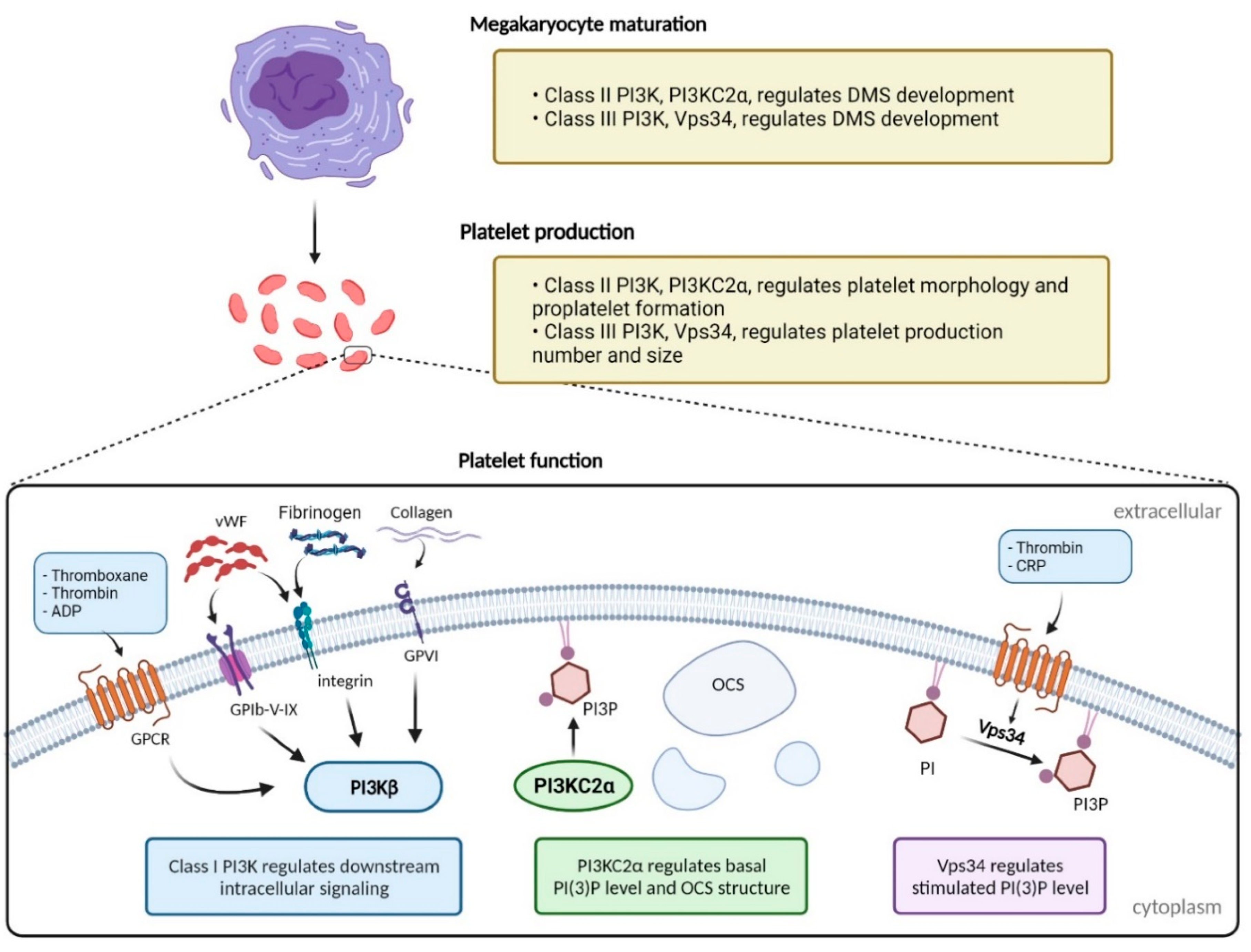
:1. Introduction
2. Phosphoinositide 3-Kinase
3. Class I PI3K
Role of Class I PI3Ks in Platelet Function and Thrombus Formation
4. Class II PI3K
Role of Class II PI3K in Platelet Function and Thrombus Formation
5. Class III PI3K
Role of Class III PI3K in Platelet Function and Thrombus Formation
6. PI3Ks as Anti-Thrombotic Targets?
7. Future Directions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Australian Bureau of Statistics. National health survey: First results, 2014–2015. In ABS Cat No 43640 55001; Australia Bureau of Statistics: Canberra, Australia, 2015. [Google Scholar]
- Heemskerk, J. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar]
- Shekhonin, B.V.; Domogatsky, S.P.; Muzykantov, V.R.; Idelson, G.L.; Rukosuev, V.S. Distribution of type I, III, IV and V collagen in normal and atherosclerotic human arterial wall: Immunomorphological characteristics. Collagen Relat. Res. 1985, 5, 355–368. [Google Scholar] [CrossRef]
- Guidetti, G.; Bertoni, A.; Viola, M.; Tira, E.; Balduini, C.; Torti, M. The small proteoglycan decorin supports adhesion and activation of human platelets. Blood 2002, 100, 1707–1714. [Google Scholar] [CrossRef]
- Bennett, J.S. Structure and function of the platelet integrin αIIbβ3. J. Clin. Investig. 2005, 115, 3363–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayes, J.; Watson, S.P.; Nieswandt, B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Investig. 2019, 129, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Durrant, T.N.; van den Bosch, M.T.; Hers, I. Integrin α(IIb)β(3) outside-in signaling. Blood 2017, 130, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, X.; Shi, X.; Zhu, M.; Wang, J.; Huang, S.; Huang, X.; Huafeng, W.; Ling, L.; Huan, D.; et al. Platelet integrin αIIbβ3: Signal transduction, regulation, and its therapeutic targeting. J. Hematol. Oncol. 2019, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Agbani, E.O.; Poole, A.W. Procoagulant platelets: Generation, function, and therapeutic targeting in thrombosis. Blood 2017, 130, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Shah, J.; Liu, S.; Yu, W. Contemporary antiplatelet therapy for secondary stroke prevention: A narrative review of current literature and guidelines. Stroke Vasc. Neurol. 2022. [Google Scholar] [CrossRef]
- COMMIT collaborative group. Addition of clopidogrel to aspirin in 45 852 patients with acute myocardial infarction: Randomised placebo-controlled trial. Lancet 2005, 366, 1607–1621. [Google Scholar]
- Palacio, S.; Hart, R.G.; Pearce, L.A.; Benavente, O.R. Effect of Addition of Clopidogrel to Aspirin on Mortality. Stroke 2012, 43, 2157–2162. [Google Scholar] [CrossRef] [PubMed]
- Degrauwe, S.; Pilgrim, T.; Aminian, A.; Noble, S.; Meier, P.; Iglesias, J.F. Dual antiplatelet therapy for secondary prevention of coronary artery disease. Open Heart 2017, 4, e000651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Kumar, P.; Prashanth, S.P.; Belagali, Y. Dual Antiplatelet Therapy in Coronary Artery Disease. Cardiol. Ther. 2020, 9, 349–361. [Google Scholar] [CrossRef]
- Capodanno, D.; Alfonso, F.; Levine, G.N.; Valgimigli, M.; Angiolillo, D.J. ACC/AHA Versus ESC Guidelines on Dual Antiplatelet Therapy: JACC Guideline Comparison. J. Am. Coll. Cardiol. 2018, 72, 2915–2931. [Google Scholar] [CrossRef]
- Capodanno, D.; Angiolillo, D.J. Aspirin for Primary Cardiovascular Risk Prevention and Beyond in Diabetes Mellitus. Circulation 2016, 134, 1579–1594. [Google Scholar] [CrossRef] [Green Version]
- Bordeaux, B.C.; Qayyum, R.; Yanek, L.R.; Vaidya, D.; Becker, L.C.; Faraday, N.; Becker, D. Effect of obesity on platelet reactivity and response to low-dose aspirin. Prev. Cardiol. 2010, 13, 56–62. [Google Scholar] [CrossRef]
- Rios, J.R.R.; Franchi, F.; Rollini, F.; Angiolillo, D.J. Diabetes and antiplatelet therapy: From bench to bedside. Cardiovasc. Diagn. Ther. 2018, 8, 594. [Google Scholar] [CrossRef]
- Kones, R.; Rumana, U. Will the epidemic of metabolic syndrome raise the prevalence of antiplatelet drug resistance? Int. J. Appl. Basic Med. Res. 2013, 3, 75–76. [Google Scholar]
- Hovens, M.M.; Snoep, J.D.; Eikenboom, J.C.; van der Bom, J.G.; Mertens, B.J.; Huisman, M.V. Prevalence of persistent platelet reactivity despite use of aspirin: A systematic review. Am. Heart J. 2007, 153, 175–181. [Google Scholar] [CrossRef]
- Liu, L.; Gao, Y.-H.; Cao, J.; Zhang, H.-X.; Fan, L.; Hu, G.-L.; Hu, Y.-X.; Li, X.-L.; Zou, X.; Li, J.-H. High prevalence of aspirin resistance in elderly patients with cardiovascular disease and metabolic syndrome. J. Geriatr. Cardiol. 2016, 13, 531–536. [Google Scholar] [PubMed]
- Kahraman, G.; Sahin, T.; Kilic, T.; Baytugan, N.Z.; Agacdiken, A.; Ural, E.; Ural, D.; Komsuoglu, B. The frequency of aspirin resistance and its risk factors in patients with metabolic syndrome. Int. J. Cardiol. 2007, 115, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Habizal, N.H.; Abdul Halim, S.; Bhaskar, S.; Wan Bebakar, W.M.; Abdullah, J.M. Prevalence of aspirin resistance in diabetic patients and its associated factors. Malays. J. Med. Sci. 2015, 22, 50–57. [Google Scholar] [PubMed]
- Angiolillo, D.J.; Shoemaker, S.B.; Desai, B.; Yuan, H.; Charlton, R.K.; Bernardo, E.; Zenni, M.; Guzman, L.; Bass, T.; Costa, M. Randomized Comparison of a High Clopidogrel Maintenance Dose in Patients With Diabetes Mellitus and Coronary Artery Disease. Circulation 2007, 115, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Meyers, R.E.; Cantley, L.C. Phosphoinositide kinases. Annu. Rev. Biochem. 1998, 67, 481–507. [Google Scholar] [CrossRef]
- Mazza, S.; Maffucci, T. Class II phosphoinositide 3-kinase C2alpha: What we learned so far. Int. J. Biochem. Mol. Biol. 2011, 2, 168. [Google Scholar]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Rudiger, W.; Parker, P.; Waterfield, M. Synthesis and Function of 3-phosphorylated inositol Lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef]
- Rathinaswamy, M.K.; Dalwadi, U.; Fleming, K.D.; Adams, C.; Stariha, J.T.B.; Pardon, E.; Baek, M.; Vadas, O.; DiMaio, F.; Steyaert, J.; et al. Structure of the phosphoinositide 3-kinase (PI3K) p110γ-p101 complex reveals molecular mechanism of GPCR activation. Sci. Adv. 2021, 7, eabj4282. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Ghigo, A.; Morello, F.; Perino, A.; Hirsch, E. Therapeutic applications of PI3K inhibitors in cardiovascular diseases. Future Med. Chem. 2013, 5, 479–492. [Google Scholar] [CrossRef]
- Maynard, J.; Emmas, S.-A.; Ble, F.-X.; Barjat, H.; Lawrie, E.; Hancox, U.; Polanska, U.; Pritchard, A.; Hudson, K. The use of 18F-Fluoro-deoxy-glucose positron emission tomography (18F-FDG PET) as a non-invasive pharmacodynamic biomarker to determine the minimally pharmacologically active dose of AZD8835, a novel PI3Kα inhibitor. PLoS ONE 2017, 12, e0183048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsolakos, N.; Durrant, T.N.; Chessa, T.; Suire, S.M.; Oxley, D.; Kulkarni, S.; Downward, J.; Perisic, O.; Williams, R.; Stephens, L.; et al. Quantitation of class IA PI3Ks in mice reveals p110-free-p85s and isoform-selective subunit associations and recruitment to receptors. Proc. Natl. Acad. Sci. USA 2018, 115, 12176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, P.; Jain, S.K. Phosphatidylinositol-3,4,5-triphosphate and cellular signaling: Implications for obesity and diabetes. Cell Physiol. Biochem. 2015, 35, 1253–1275. [Google Scholar] [CrossRef] [PubMed]
- Riehle, R.D.; Cornea, S.; Degterev, A. Role of phosphatidylinositol 3, 4, 5-trisphosphate in cell signaling. Lipid-Mediat. Protein Signal. 2013, 991, 105–139. [Google Scholar]
- Hemmati, S.; Sinclair, T.; Tong, M.; Bartholdy, B.; Okabe, R.O.; Ames, K.; Ostrodka, L.; Haque, T.; Kaur, I.; Mills, T.; et al. PI3 kinase alpha and delta promote hematopoietic stem cell activation. JCI Insight 2019, 5, e125832. [Google Scholar] [CrossRef]
- Kok, K.; Nock, G.E.; Verrall, E.A.G.; Mitchell, M.P.; Hommes, D.W.; Peppelenbosch, M.P.; Vanhaesebroeck, B. Regulation of p110δ PI 3-Kinase Gene Expression. PLoS ONE 2009, 4, e5145. [Google Scholar] [CrossRef]
- Triscott, J.; Rubin, M.A. Prostate Power Play: Does Pik3ca Accelerate Pten-Deficient Cancer Progression? Cancer Discov. 2018, 8, 682–685. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.; Riggins, G. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-Y.; Wei, S.-J.; Lin, Y.-C.; Lung, J.-C.; Chang, T.-C.; Whang-Peng, J.; Liu, J.; Yang, D.; Yang, W.; Shen, C.-Y. PIK3CA as an oncogene in cervical cancer. Oncogene 2000, 19, 2739. [Google Scholar] [CrossRef] [Green Version]
- Hervieu, A.; Kermorgant, S. The Role of PI3K in Met Driven Cancer: A Recap. Front. Mol. Biosci. 2018, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.M.; Kalifa, S.; Das, J.R.; Bhatti, T.; Gay, M.; Williams, D.; Taliferro-Smith, L.; De-Marzo, A. The role of PI 3-kinase p110β in AKT signally, cell survival, and proliferation in human prostate cancer cells. Prostate 2010, 70, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.; Salpekar, A.; Waterfield, M. Impaired B and T cell antigen receptor signaling in p110δ PI 3-kinase mutant mice. Science 2002, 297, 1031–1034. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.; Bardi, G.; Bell, S.E.; Chantry, D.; Downes, C.P.; Gray, A.; Humphries, L.; Rawlings, D.; Reynolds, H.; Vigorito, E. A crucial role for the p110δ subunit of phosphatidylinositol 3-kinase in B cell development and activation. J. Exp. Med. 2002, 196, 753–763. [Google Scholar] [CrossRef]
- Okkenhaug, K. Signaling by the Phosphoinositide 3-Kinase Family in Immune Cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [Green Version]
- Ali, K.; Bilancio, A.; Thomas, M.; Pearce, W.; Gilfillan, A.M.; Tkaczyk, C.; Kuehn, N.; Gray, A.; Giddings, J.; Peskett, E. Essential role for the p110δ phosphoinositide 3-kinase in the allergic response. Nature 2004, 431, 1007. [Google Scholar] [CrossRef]
- Barber, D.F.; Bartolomé, A.; Hernandez, C.; Flores, J.M.; Redondo, C.; Fernandez-Arias, C.; Camps, M.; Rückle, T.; Schwarz, M.K.; Rodríguez, S.; et al. PI3Kγ inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat. Med. 2005, 11, 933–935. [Google Scholar] [CrossRef]
- Camps, M.; Rückle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Francon, B. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Valet, C.; Severin, S.; Chicanne, G.; Laurent, P.A.; Gaits-Iacovoni, F.; Gratacap, M.-P.; Payrastre, B. The role of class I, II and III PI 3-kinases in platelet production and activation and their implication in thrombosis. Adv. Biol. Regul. 2016, 61, 33–41. [Google Scholar] [CrossRef]
- Martin, V.; Guillermet-Guibert, J.; Chicanne, G.; Cabou, C.; Jandrot-Perrus, M.; Plantavid, M.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.-P. Deletion of the p110β isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood 2010, 115, 2008–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P.; Schoenwaelder, S.M.; Goncalves, I.; Nesbitt, W.S.; Yap, C.L.; Wright, C.E.; Kenche, V.; Anderson, K.; Dopheide, S.; Yuan, Y. PI 3-kinase p110β: A new target for antithrombotic therapy. Nat. Med. 2005, 11, 507–514. [Google Scholar] [CrossRef]
- Nylander, S.; Kull, B.; Björkman, J.; Ulvinge, J.; Oakes, N.; Emanuelsson, B.; Andersson, M.; Skarby, T.; Inghardt, T.; Fjellstrom, O. Human target validation of phosphoinositide 3-kinase (PI3K) β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Laurent, P.-A.; Séverin, S.; Hechler, B.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.-P. Platelet PI3Kβ and GSK3 regulate thrombus stability at a high shear rate. Blood 2015, 125, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, T.A.; Moore, S.F.; Williams, C.M.; Poole, A.W.; Vanhaesebroeck, B.; Hers, I. Phosphoinositide 3-kinases p110α and p110β have differential roles in insulin-like growth factor-1–mediated Akt phosphorylation and platelet priming. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1681–1688. [Google Scholar] [CrossRef] [Green Version]
- Laurent, P.-A.; Hechler, B.; Solinhac, R.; Ragab, A.; Cabou, C.; Anquetil, T.; Severin, S.; Denis, C.; Mangin, P.; Vanhaesebroeck, B. Impact of PI3Kα (phosphoinositide 3-kinase alpha) inhibition on hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2041–2053. [Google Scholar] [CrossRef] [Green Version]
- Barrachina, M.N.; Izquierdo, I.; Hermida-Nogueira, L.; Morán, L.A.; Pérez, A.; Arroyo, A.B.; Garcia-Barbera, N.; Gonzalez-Conejero, R.; Troitino, S.; Eble, J.; et al. The PI3Kδ Inhibitor Idelalisib Diminishes Platelet Function and Shows Antithrombotic Potential. Int. J. Mol. Sci. 2021, 22, 3304. [Google Scholar] [CrossRef]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.; Kelkar, D.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Rittenhouse, S.E. Phosphoinositide 3-kinase activation and platelet function. Blood 1996, 88, 4401–4414. [Google Scholar] [CrossRef] [Green Version]
- Bird, J.E.; Smith, P.L.; Bostwick, J.S.; Shipkova, P.; Schumacher, W.A. Bleeding response induced by anti-thrombotic doses of a phosphoinositide 3-kinase (PI3K)-β inhibitor in mice. Thromb. Res. 2011, 127, 560–564. [Google Scholar] [CrossRef]
- Nylander, S.; Wågberg, F.; Andersson, M.; Skärby, T.; Gustafsson, D. Exploration of efficacy and bleeding with combined phosphoinositide 3-kinase β inhibition and aspirin in man. J. Thromb. Haemost. 2015, 13, 1494–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senis, Y.A.; Atkinson, B.T.; Pearce, A.C.; Wonerow, P.; Auger, J.M.; Okkenhaug, K.; Pearce, W.; Vigorito, E.; Vanhaesebroeck, B.; Turner, M.; et al. Role of the p110delta PI 3-kinase in integrin and ITAM receptor signalling in platelets. Platelets 2005, 16, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Bosco, O.; Tropel, P.; Laffargue, M.; Calvez, R.; Altruda, F.; Wymann, M.; Montrucchio, G. Resistance to thromboembolism in PI3Kγ-deficient mice. FASEB J. 2001, 15, 2019–2021. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.; Wang, Y.; Draznin, J.; Eslin, D.; Bennett, J.S.; Poncz, M.; Wu, D.; Abrams, C. The relative role of PLCbeta and PI3Kgamma in platelet activation. Blood 2005, 106, 110–117. [Google Scholar] [CrossRef]
- Cosemans, J.M.E.M.; Munnix, I.C.A.; Wetzker, R.; Heller, R.; Jackson, S.P.; Heemskerk, J.W.M. Continuous signaling via PI3K isoforms β and γ is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood 2006, 108, 3045–3052. [Google Scholar] [CrossRef]
- Schoenwaelder, S.M.; Ono, A.; Sturgeon, S.; Chan, S.M.; Mangin, P.; Maxwell, M.J.; Turnbull, S.; Mulchandani, M.; Anderson, K.; Kauffenstein, G. Identification of a unique co-operative phosphoinositide 3-kinase signaling mechanism regulating integrin αIIbβ3 adhesive function in platelets. J. Biol. Chem. 2007, 282, 28648–28658. [Google Scholar] [CrossRef] [Green Version]
- MacDougall, L.K.; Domin, J.; Waterfield, M.D. A family of phosphoinositide 3-kinases in Drosophila identifies a new mediator of signal transduction. Curr. Biol. 1995, 5, 1404–1415. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Banfic, H.; Straforini, F.; Tosi, L.; Volinia, S.; Rittenhouse, S.E. A Type II Phosphoinositide 3-Kinase Is Stimulated via Activated Integrin in Platelets: A Source of Phosphatidylinositol 3-Phosphate. J. Biol. Chem. 1998, 273, 14081–14084. [Google Scholar] [CrossRef] [Green Version]
- Falasca, M.; Maffucci, T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem. J. 2012, 443, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Margaria, J.P.; Ratto, E.; Gozzelino, L.; Li, H.; Hirsch, E. Class II PI3Ks at the Intersection between Signal Transduction and Membrane Trafficking. Biomolecules 2019, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Marat, A.L.; Wallroth, A.; Lo, W.-T.; Müller, R.; Norata, G.D.; Falasca, M.; Schultz, C.; Haucke, V. mTORC1 activity repression by late endosomal phosphatidylinositol 3, 4-bisphosphate. Science 2017, 356, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Hamilton, J.R.; Selvadurai, M.; Sundaram, K.; Adamska, A.; Thompson, P.E. Class II Phosphoinositide 3-Kinases as Novel Drug Targets: Miniperspective. J. Med. Chem. 2016, 60, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Elis, W.; Triantafellow, E.; Wolters, N.M.; Sian, K.R.; Caponigro, G.; Borawski, J.; Gaither, L.; Murphy, L.; Finan, P.; MacKeigan, J. Down-regulation of class II phosphoinositide 3-kinase α expression below a critical threshold induces apoptotic cell death. Mol. Cancer Res. 2008, 6, 614–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, D.P.; Vogel, P.; Wims, M.; Moberg, K.; Humphries, J.; Jhaver, K.G.; DaCosta, C.; Shadoan, M.; Xu, N.; Hansen, G.; et al. Requirement for Class II Phosphoinositide 3-Kinase C2α in Maintenance of Glomerular Structure and Function. Mol. Cell. Biol. 2011, 31, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiosano, D.; Baris, H.N.; Chen, A.; Hitzert, M.M.; Schueler, M.; Gulluni, F.; Wiesener, A.; Bergua, A.; Mory, A.; Copeland, B. Mutations in PIK3C2A cause syndromic short stature, skeletal abnormalities, and cataracts associated with ciliary dysfunction. PLoS Genet. 2019, 15, e1008088. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K.; Yoshida, K.; Cui, H.; Wakayama, T.; Takuwa, N.; Okamoto, Y.; Du, W.; Qi, X.; Asanuma, K.; Sugihara, K.; et al. Endothelial PI3K-C2[alpha], a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat. Med. 2012, 18, 1560–1569. [Google Scholar] [CrossRef] [Green Version]
- Franco, I.; Gulluni, F.; Campa Carlo, C.; Costa, C.; Margaria Jean, P.; Ciraolo, E.; Martini, M.; Monteyne, D.; DeLuca, E.; Germena, G.; et al. PI3K Class II α Controls Spatially Restricted Endosomal PtdIns3P and Rab11 Activation to Promote Primary Cilium Function. Dev. Cell 2014, 28, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Alliouachene, S.; Bilanges, B.; Chicanne, G.; Anderson, K.E.; Pearce, W.; Ali, K.; Valet, C.; Posor, Y.; Low, P.; Chaussade, C. Inactivation of the class II PI3K-C2β potentiates insulin signaling and sensitivity. Cell Rep. 2015, 13, 1881–1894. [Google Scholar] [CrossRef] [Green Version]
- Kolic, J.; Fox, J.E.M.; Chepurny, O.G.; Spigelman, A.F.; Ferdaoussi, M.; Schwede, F.; Holz, G.; MacDonald, P. PI3 kinases p110a and PI3K-C2b negatively regulate cAMP via PDE3/8 to control insulin secretion in mouse and human islets. Mol. Metab. 2016, 5, 459–471. [Google Scholar] [CrossRef]
- Gratacap, M.P.; Darcourt, J.; Vanhaesebroeck, B.; Chicanne, G.; Payrastre, B.; Larrue, V.; Solinhac, R.; Jaffre, A.; Vivien, D.; Anquetil, T. Use of PI3KC2B Inhibitors for the Preservation of Vascular Endothelial Cell Barrier Integrity. US Patent US20210238605A1, 5 August 2021. [Google Scholar]
- El Sheikh, S.S.; Domin, J.; Tomtitchong, P.; Abel, P.; Stamp, G.; Lalani, E.-N. Topographical expression of class IA and class II phosphoinositide 3-kinase enzymes in normal human tissues is consistent with a role in differentiation. BMC Clin. Pathol. 2003, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Mountford, J.K.; Petitjean, C.; Putra, H.W.K.; McCafferty, J.A.; Setiabakti, N.M.; Lee, H.; Tonnesen, L.; McFadyen, J.; Schoenwaelder, S.; Eckly, A. The class II PI 3-kinase, PI3KC2α, links platelet internal membrane structure to shear-dependent adhesive function. Nat. Commun. 2015, 6, 6535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valet, C.; Chicanne, G.; Severac, C.; Chaussade, C.; Whitehead, M.A.; Cabou, C.; Gratacap, M.-P.; Gaits-Iacovoni, F.; Vanhaesebroeck, B.; Payrastre, B. Essential role of class II PI3K-C2α in platelet membrane morphology. Blood 2015, 126, 1128–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvadurai, M.V.; Hamilton, J.R. Structure and function of the open canalicular system—The platelet’s specialized internal membrane network. Platelets 2018, 29, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Selvadurai, M.V.; Brazilek, R.J.; Moon, M.J.; Rinckel, J.Y.; Eckly, A.; Gachet, C.; Meikle, P.; Nandurkar, H.; Nesbitt, W.; Hamilton, J. The PI 3-kinase PI 3 KC 2α regulates mouse platelet membrane structure and function independently of membrane lipid composition. FEBS Lett. 2019, 593, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvadurai, M.V.; Moon, M.J.; Mountford, S.J.; Ma, X.; Zheng, Z.; Jennings, I.G.; Setiabakti, N.; Iman, R.; Brazilek, R.; Abidin, N.; et al. Disrupting the platelet internal membrane via PI3KC2α inhibition impairs thrombosis independently of canonical platelet activation. Sci. Transl. Med. 2020, 12, eaar8430. [Google Scholar] [CrossRef]
- Petitjean, C.; Setiabakti, N.M.; Mountford, J.K.; Arthur, J.F.; Ellis, S.; Hamilton, J.R. Combined deficiency of PI3KC2α and PI3KC2β reveals a nonredundant role for PI3KC2α in regulating mouse platelet structure and thrombus stability. Platelets 2016, 27, 402–409. [Google Scholar] [CrossRef]
- Herman, P.K.; Emr, S.D. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. Mol. Cell. Biol. 1990, 10, 6742–6754. [Google Scholar]
- Jaber, N.; Zong, W.-X. Class III PI3K Vps34: Essential roles in autophagy, endocytosis, and heart and liver function. Ann. N. Y. Acad. Sci. 2013, 1280, 48. [Google Scholar] [CrossRef]
- McLeod, I.X.; Zhou, X.; Li, Q.-J.; Wang, F.; He, Y.-W. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Rα surface expression. J. Immunol. 2011, 187, 5051–5061. [Google Scholar] [CrossRef] [Green Version]
- Bilanges, B.; Alliouachene, S.; Pearce, W.; Morelli, D.; Szabadkai, G.; Chung, Y.-L.; Chicanne, G.; Valet, C.; Hill, J.; Voshol, P.; et al. Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat. Commun. 2017, 8, 1804. [Google Scholar] [CrossRef]
- Saito, T.; Aghalar, M.R.; Lachman, H.M. Analysis of PIK3C3 promoter variant in African-Americans with schizophrenia. Schizophr. Res. 2005, 76, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Stopkova, P.; Saito, T.; Papolos, D.F.; Vevera, J.; Paclt, I.; Zukov, I.; Bersson, Y.; Margolis, B.; Strous, R.; Lachma, H. Identification of PIK3C3 promoter variant associated with bipolar disorder and schizophrenia. Biol. Psychiatry 2004, 55, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Grieco, G.; Janssens, V.; Gaide Chevronnay, H.P.; N’Kuli, F.; Van Der Smissen, P.; Wang, T.; Shan, J.; Vainio, S.; Bilanges, B.; Jouret, F.; et al. Vps34/PI3KC3 deletion in kidney proximal tubules impairs apical trafficking and blocks autophagic flux, causing a Fanconi-like syndrome and renal insufficiency. Sci. Rep. 2018, 8, 14133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valet, C.; Levade, M.; Chicanne, G.; Bilanges, B.; Cabou, C.; Viaud, J.; Gratacap, M.-P.; Gaits-Iacovoni, F.; Vanhaesebroeck, B.; Payrastre, B. A dual role for the class III PI3K, Vps34, in platelet production and thrombus growth. Blood 2017, 130, 2032–2042. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hu, M.; Luo, D.; Yue, M.; Wang, S.; Chen, X.; Zhou, Y.; Wang, Y.; Cai, Y.; Hu, X.; et al. Class III PI3K Positively Regulates Platelet Activation and Thrombosis via PI(3)P-Directed Function of NADPH Oxidase. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2075–2086. [Google Scholar] [CrossRef] [Green Version]
- Bertović, I.; Kurelić, R.; Milošević, I.; Bender, M.; Krauss, M.; Haucke, V.; Jurak Begonja, A. Vps34 derived phosphatidylinositol 3-monophosphate modulates megakaryocyte maturation and proplatelet production through late endosomes/lysosomes. J. Thromb. Haemost. 2020, 18, 1756–1772. [Google Scholar] [CrossRef]
- Marquis-Gravel, G.; Roe, M.T.; Harrington, R.A.; Muñoz, D.; Hernandez, A.F.; Jones, W.S. Revisiting the Role of Aspirin for the Primary Prevention of Cardiovascular Disease. Circulation 2019, 140, 1115–1124. [Google Scholar] [CrossRef]
- McNeil, J.J.; Wolfe, R.; Woods, R.L.; Tonkin, A.M.; Donnan, G.A.; Nelson, M.R.; Reid, C.; Lockery, J.E.; Kirpach, B.; Storey, E.; et al. Effect of Aspirin on Cardiovascular Events and Bleeding in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1509–1518. [Google Scholar] [CrossRef]
- Welsh, J.D.; Stalker, T.J.; Voronov, R.; Muthard, R.W.; Tomaiuolo, M.; Diamond, S.L.; Brass, L.F. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood 2014, 124, 1808–1815. [Google Scholar] [CrossRef]
- Torti, M. PI3Kβ inhibition: All that glitters is not gold. Blood J. Am. Soc. Hematol. 2015, 125, 750–751. [Google Scholar] [CrossRef] [Green Version]
- Watala, C.; Golanski, J.; Pluta, J.; Boncler, M.; Rozalski, M.; Luzak, B.; Kropiwnicka, A.; Drzewoski, J. Reduced sensitivity of platelets from type 2 diabetic patients to acetylsalicylic acid (aspirin)—its relation to metabolic control. Thromb. Res. 2004, 113, 101–113. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| PI3K | Species/Model | Platelet/Thrombus Readout | References | |||
|---|---|---|---|---|---|---|
| Platelet Function | Thrombus Formation | |||||
| Ex Vivo * | In Vivo ** | |||||
| Thrombosis | Bleeding Time | |||||
| Class I PI3K—generates PI(3,4,5)P3 | ||||||
| PI3Kα | Mouse genetic | ↓ | - | ↓ | - | [56,57] |
| Human—PI3Kα inhibitor | ↓ | - | n.d. | n.d. | [57,58] | |
| PI3Kβ | Mouse genetic | ↓ | U/↓ | ↓ | - | [52,55] |
| Human—PI3Kβ inhibitor | ↓ | U | n.d. | ↑ | [55,58] | |
| PI3Kδ | Mouse genetic | ↓ | - | n.d. | n.d. | [63] |
| Human—PI3Kδ inhibitor | ↓ | n.d. | n.d. | n.d. | [58] | |
| PI3Kγ | Mouse genetic | ↓ | U | ↓ | - | [64,65,66] |
| Class II PI3K—generates PI(3)P and PI(3,4)P2 | ||||||
| PI3KC2α | Mouse genetic | - | U | ↓ | - | [83,84] |
| Mouse wild-type with PI3KC2α inhibitor | n.d. | n.d. | ↓ | - | [87] | |
| Human—PI3KC2α inhibitor | - | ↓ | n.d. | n.d. | [87] | |
| Human genetic—homozygous loss-of-function of PI3KC2α | No thrombotic phenotype reported Developed novel syndrome—dysmorphic facial features, short statures, cataracts, multiple skeletal and neurological abnormalities | [76] | ||||
| PI3KC2β | Mouse genetic | - | - | - | - | [83] |
| Class III PI3K—generates PI(3)P | ||||||
| Vps34 | Mouse genetic | n.d. | ↓ | ↓ | - | [96,97] |
| Human—Vps34 inhibitor | ↓ | ↓ | n.d. | n.d. | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setiabakti, N.M.; Larsson, P.; Hamilton, J.R. Phosphoinositide 3-Kinases as Potential Targets for Thrombosis Prevention. Int. J. Mol. Sci. 2022, 23, 4840. https://doi.org/10.3390/ijms23094840
Setiabakti NM, Larsson P, Hamilton JR. Phosphoinositide 3-Kinases as Potential Targets for Thrombosis Prevention. International Journal of Molecular Sciences. 2022; 23(9):4840. https://doi.org/10.3390/ijms23094840
Chicago/Turabian StyleSetiabakti, Natasha M., Pia Larsson, and Justin R. Hamilton. 2022. "Phosphoinositide 3-Kinases as Potential Targets for Thrombosis Prevention" International Journal of Molecular Sciences 23, no. 9: 4840. https://doi.org/10.3390/ijms23094840