
Recent Experimental Studies of Maternal Obesity, Diabetes during Pregnancy and the Developmental Origins of Cardiovascular Disease


{kind=link}
Abstract
:1. Introduction
2. Cardiovascular Disease and Diabetes
3. Youth Onset Type 2 Diabetes and Cardiovascular Disease
4. The Developmental Origins of Health and Disease Theory
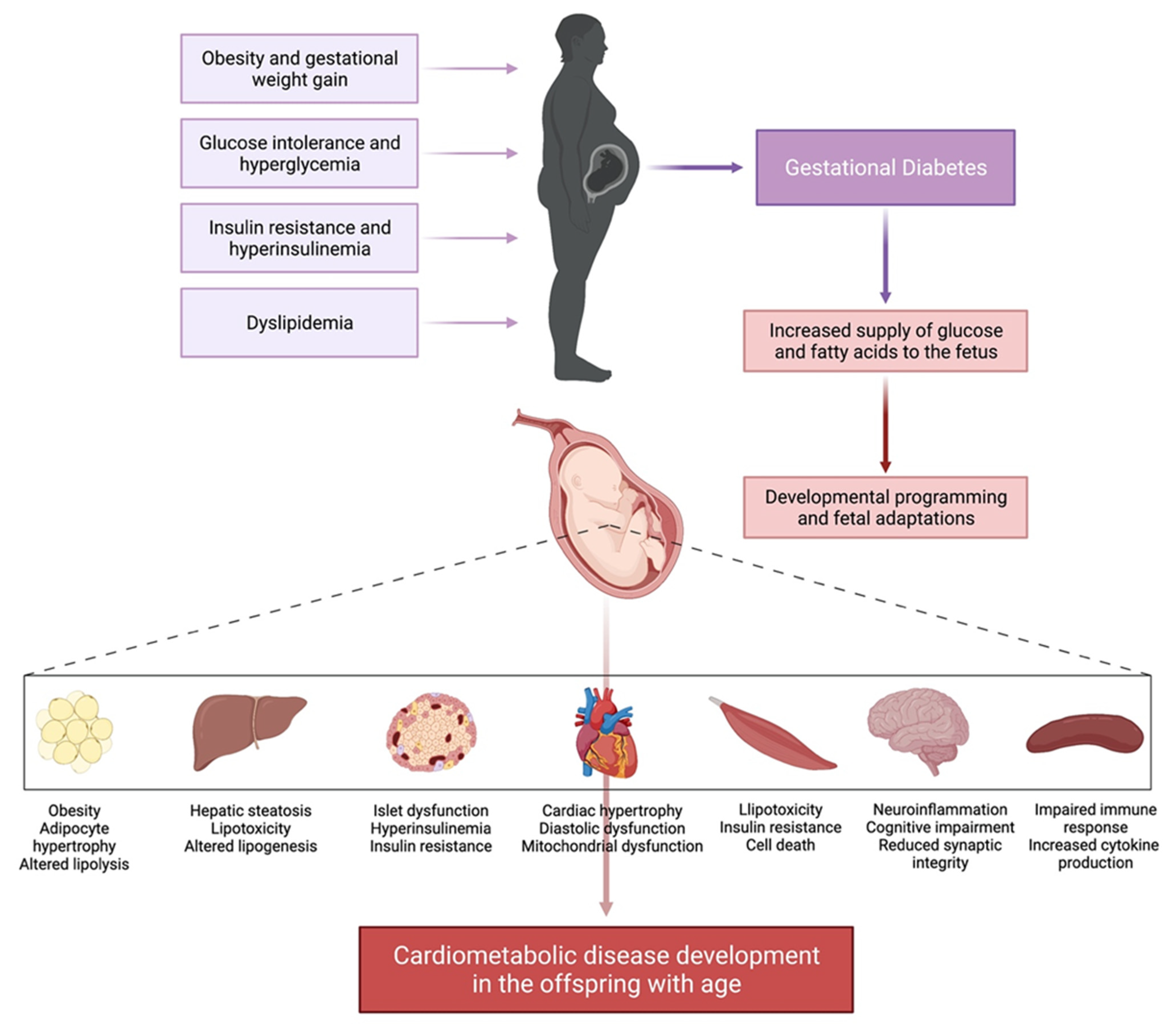
5. Maternal Obesity and Gestational Diabetes Mellitus
6. Animal Models of Maternal Obesity and Diabetes in Pregnancy
6.1. Metabolic Disease Development in Offspring Exposed to Diabetes during Pregnancy and Maternal Obesity
6.2. The Effects of Maternal Obesity on Heart Disease Development in The Offspring
6.3. The Effects of Diabetes during Pregnancy on Heart Disease Development in the Offspring
6.4. The Effects of Maternal Obesity on Vascular Function in The Offspring
7. Interventions for the Developmental Origins of Cardiovascular Disease
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Public Health Agency of Canada. Tracking Heart Disease and Stroke in Canada. 2009. Available online: https://www.phac-aspc.gc.ca/publicat/2009/cvd-avc/pdf/cvd-avs-2009-eng.pdf (accessed on 15 March 2022).
- Mahmood, S.S.; Levy, D.; Vasan, R.S.; Wang, T.J. The Framingham Heart Study and the epidemiology of cardiovascular disease: A historical perspective. Lancet 2014, 383, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Andersson, C.; Johnson, A.D.; Benjamin, E.J.; Levy, D.; Vasan, R.S. 70-year legacy of the Framingham Heart Study. Nat. Rev. Cardiol. 2019, 16, 687–698. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; Available online: http://www.diabetesatlas.org (accessed on 15 March 2022).
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Orchard, T.J.; Costacou, T.; Kretowski, A.; Nesto, R.W. Type 1 diabetes and coronary artery disease. Diabetes Care 2006, 29, 2528–2538. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003, 289, 76–79. [Google Scholar] [CrossRef]
- Kannel, W.B.; Dawber, T.R.; Kagan, A.; Revotskie, N.; Stokes, J. 3rd, Factors of risk in the development of coronary heart disease—Six year follow-up experience. The Framingham Study. Ann. Intern. Med. 1961, 55, 33–50. [Google Scholar] [CrossRef]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The Framingham Study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular risk factors: The Framingham study. Circulation 1979, 59, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.W.; D’Agostino, R.B.; Sullivan, L.; Parise, H.; Kannel, W.B. Overweight and obesity as determinants of cardiovascular risk: The Framingham experience. Arch. Intern. Med. 2002, 162, 1867–1872. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.W.; D’Agostino, R.B.; Levy, D.; Belanger, A.M.; Silbershatz, H.; Kannel, W.B. Prediction of coronary heart disease using risk factor categories. Circulation 1998, 97, 1837–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.J.; McNamara, P.M.; Gordon, T.; Kannel, W.B. Morbidity and mortality in diabetics in the Framingham population. Sixteen year follow-up study. Diabetes 1974, 23, 105–111. [Google Scholar] [CrossRef]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef]
- Fox, C.S.; Coady, S.; Sorlie, P.D.; D’Agostino, R.B.; Pencina, M.J.; Vasan, R.S.; Meigs, J.B.; Levy, D.; Savage, P.J. Increasing cardiovascular disease burden due to diabetes mellitus: The Framingham Heart Study. Circulation 2007, 115, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Constantino, M.I.; Molyneaux, L.; Limacher-Gisler, F.; Al-Saeed, A.; Luo, C.; Wu, T.; Twigg, S.M.; Yue, D.K.; Wong, J. Long-term complications and mortality in young-onset diabetes: Type 2 diabetes is more hazardous and lethal than type 1 diabetes. Diabetes Care 2013, 36, 3863–3869. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Yaniv, G.; Levine, H.; Leiba, A.; Goldberger, N.; Derazne, E.; Ben-Ami Shor, D.; Tzur, D.; Afek, A.; Shamiss, A.; et al. Body-Mass Index in 2.3 Million Adolescents and Cardiovascular Death in Adulthood. N. Engl. J. Med. 2016, 374, 2430–2440. [Google Scholar] [CrossRef]
- Levitt Katz, L.; Gidding, S.S.; Bacha, F.; Hirst, K.; McKay, S.; Pyle, L.; Lima, J.A.; Group, T.S. Alterations in left ventricular, left atrial, and right ventricular structure and function to cardiovascular risk factors in adolescents with type 2 diabetes participating in the TODAY clinical trial. Pediatr. Diabetes 2015, 16, 39–47. [Google Scholar]
- Crowley, D.I.; Khoury, P.R.; Urbina, E.M.; Ippisch, H.M.; Kimball, T.R. Cardiovascular impact of the pediatric obesity epidemic: Higher left ventricular mass is related to higher body mass index. J. Pediatr. 2011, 158, 709–714.e1. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, W.; Srinivasan, S.R.; Bond, M.G.; Tang, R.; Urbina, E.M.; Berenson, G.S. Childhood cardiovascular risk factors and carotid vascular changes in adulthood: The Bogalusa Heart Study. JAMA 2003, 290, 2271–2276. [Google Scholar] [CrossRef]
- Juonala, M.; Magnussen, C.G.; Berenson, G.S.; Venn, A.; Burns, T.L.; Sabin, M.A.; Srinivasan, S.R.; Daniels, S.R.; Davis, P.H.; Chen, W.; et al. Childhood adiposity, adult adiposity, and cardiovascular risk factors. N. Engl. J. Med. 2011, 365, 1876–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raitakari, O.T.; Juonala, M.; Kahonen, M.; Taittonen, L.; Laitinen, T.; Maki-Torkko, N.; Jarvisalo, M.J.; Uhari, M.; Jokinen, E.; Ronnemaa, T.; et al. Cardiovascular risk factors in childhood and carotid artery intima-media thickness in adulthood: The Cardiovascular Risk in Young Finns Study. JAMA 2003, 290, 2277–2283. [Google Scholar] [CrossRef]
- Urbina, E.M.; Gidding, S.S.; Bao, W.; Pickoff, A.S.; Berdusis, K.; Berenson, G.S. Effect of body size, ponderosity, and blood pressure on left ventricular growth in children and young adults in the Bogalusa Heart Study. Circulation 1995, 91, 2400–2406. [Google Scholar] [CrossRef] [PubMed]
- McKee, P.A.; Castelli, W.P.; McNamara, P.M.; Kannel, W.B. The natural history of congestive heart failure: The Framingham study. N. Engl. J. Med. 1971, 285, 1441–1446. [Google Scholar] [CrossRef]
- Feinleib, M.; Kannel, W.B.; Garrison, R.J.; McNamara, P.M.; Castelli, W.P. The framingham offspring study. Design and preliminary data. Prev. Med. 1975, 4, 518–525. [Google Scholar] [CrossRef]
- Splansky, G.L.; Corey, D.; Yang, Q.; Atwood, L.D.; Cupples, L.A.; Benjamin, E.J.; D’Agostino, R.B.; Fox, C.S.; Larson, M.G.; Murabito, J.M.; et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: Design, recruitment, and initial examination. Am. J. Epidemiol. 2007, 165, 1328–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, R.H.; Kiely, D.K.; Cupples, L.A.; Kannel, W.B. Parental history is an independent risk factor for coronary artery disease: The Framingham Study. Am. Heart J. 1990, 120, 963–969. [Google Scholar] [CrossRef]
- Lee, D.S.; Pencina, M.J.; Benjamin, E.J.; Wang, T.J.; Levy, D.; O’Donnell, C.J.; Nam, B.H.; Larson, M.G.; D’Agostino, R.B.; Vasan, R.S. Association of parental heart failure with risk of heart failure in offspring. N. Engl. J. Med. 2006, 355, 138–147. [Google Scholar] [CrossRef]
- Meigs, J.B.; Cupples, L.A.; Wilson, P.W. Parental transmission of type 2 diabetes: The Framingham Offspring Study. Diabetes 2000, 49, 2201–2207. [Google Scholar] [CrossRef] [Green Version]
- Mendelson, M.M.; Lyass, A.; O’Donnell, C.J.; D’Agostino, R.B.; Levy, D. Association of Maternal Prepregnancy Dyslipidemia With Adult Offspring Dyslipidemia in Excess of Anthropometric, Lifestyle, and Genetic Factors in the Framingham Heart Study. JAMA Cardiol. 2016, 1, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, M.; Wang, Z.; Bassel-Duby, R.; Olson, E.N. Genetic and epigenetic regulation of cardiomyocytes in development, regeneration and disease. Development 2018, 145, dev171983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluckman, P.D.; Hanson, M.A.; Cooper, C.; Thornburg, K.L. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 2008, 359, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit. Rev. Clin. Lab. Sci. 2018, 55, 71–101. [Google Scholar] [CrossRef]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef]
- Barker, D.J. Maternal nutrition, fetal nutrition, and disease in later life. Nutrition 1997, 13, 807–813. [Google Scholar] [CrossRef]
- Barker, D.J.; Godfrey, K.M.; Gluckman, P.D.; Harding, J.E.; Owens, J.A.; Robinson, J.S. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993, 341, 938–941. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C.; Forsen, T.J.; Kajantie, E.; Eriksson, J.G. Trajectories of growth among children who have coronary events as adults. N. Engl. J. Med. 2005, 353, 1802–1809. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C.; Winter, P.D.; Margetts, B.; Simmonds, S.J. Weight in Infancy and Death from Ischaemic Heart Disease. Lancet 1989, 334, 577–580. [Google Scholar] [CrossRef]
- Kereliuk, S.M.; Brawerman, G.M.; Dolinsky, V.W. Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring. Int. J. Mol. Sci. 2017, 18, 1451. [Google Scholar] [CrossRef] [PubMed]
- Talbot, C.P.J.; Dolinsky, V.W. Sex differences in the developmental origins of cardiometabolic disease following exposure to maternal obesity and gestational diabetes. Appl. Physiol. Nutr. Metab. 2019, 44, 687–695. [Google Scholar] [CrossRef] [PubMed]
- McNamara, B.J.; Gubhaju, L.; Chamberlain, C.; Stanley, F.; Eades, S.J. Early life influences on cardio-metabolic disease risk in aboriginal populations—What is the evidence? A systematic review of longitudinal and case-control studies. Int. J. Epidemiol. 2012, 41, 1661–1682. [Google Scholar] [CrossRef]
- Huang, R.C.; Prescott, S.L.; Godfrey, K.M.; Davis, E.A. Assessment of cardiometabolic risk in children in population studies: Underpinning developmental origins of health and disease mother-offspring cohort studies. J. Nutr. Sci. 2015, 4, e12. [Google Scholar] [CrossRef] [Green Version]
- Catalano, P.M.; Huston, L.; Amini, S.B.; Kalhan, S.C. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am. J. Obstet. Gynecol. 1999, 180, 903–916. [Google Scholar] [CrossRef]
- Flegal, K.M.; Kruszon-Moran, D.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA 2016, 315, 2284–2291. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, E.P.; Quesenberry, C.P., Jr.; Jacobs, D.R., Jr.; Feng, J.; Lewis, C.E.; Sidney, S. Longitudinal study of prepregnancy cardiometabolic risk factors and subsequent risk of gestational diabetes mellitus: The CARDIA study. Am. J. Epidemiol. 2010, 172, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, L.; Pettitt, D.J. Gestational diabetes mellitus. JAMA 2001, 286, 2516–2518. [Google Scholar] [CrossRef]
- Ferrara, A. Increasing prevalence of gestational diabetes mellitus: A public health perspective. Diabetes Care 2007, 30, S141–S146. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, T.A.; Xiang, A.; Kjos, S.L.; Watanabe, R. What is gestational diabetes? Diabetes Care 2007, 30, S105–S111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeting, A.; Wong, J.; Murphy, H.R.; Ross, G.P. A clinical update on Gestational Diabetes Mellitus. Endocr. Rev. 2022, bnac003. [Google Scholar] [CrossRef] [PubMed]
- Moyce, B.L.; Dolinsky, V.W. Maternal beta-cell adaptations in pregnancy and placental signalling: Implications for gestational diabetes. Int. J. Mol. Sci. 2018, 19, 3467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellamy, L.; Casas, J.-P.; Hingorani, A.D.; Williams, D. Type 2 diabetes mellitus after gestational diabetes: A systematic review and meta-analysis. Lancet 2009, 373, 1773–1779. [Google Scholar] [CrossRef]
- Feig, D.S.; Zinman, B.; Wang, X.; Hux, J.E. Risk of development of diabetes mellitus after diagnosis of gestational diabetes. CMAJ 2008, 179, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, E.P.; Lewis, C.E.; Tsai, A.L.; Chiang, V.; Carnethon, M.; Quesenberry, C.P., Jr.; Sidney, S. A 20-year prospective study of childbearing and incidence of diabetes in young women, controlling for glycemia before conception: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Diabetes 2007, 56, 2990–2996. [Google Scholar] [CrossRef] [Green Version]
- Carr, D.B.; Utzschneider, K.M.; Hull, R.L.; Tong, J.; Wallace, T.M.; Kodama, K.; Shofer, J.B.; Heckbert, S.R.; Boyko, E.J.; Fujimoto, W.Y.; et al. Gestational diabetes mellitus increases the risk of cardiovascular disease in women with a family history of type 2 diabetes. Diabetes Care 2006, 29, 2078–2083. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, E.P.; Chiang, V.; Pletcher, M.J.; Jacobs, D.R.; Quesenberry, C.P.; Sidney, S.; Lewis, C.E. History of gestational diabetes mellitus and future risk of atherosclerosis in mid-life: The Coronary Artery Risk Development in Young Adults study. J. Am. Heart Assoc. 2014, 3, e000490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandi, S.M.; Filion, K.B.; Yoon, S.; Ayele, H.T.; Doyle, C.M.; Hutcheon, J.A.; Smith, G.N.; Gore, G.C.; Ray, J.G.; Nerenberg, K.; et al. Cardiovascular Disease-Related Morbidity and Mortality in Women With a History of Pregnancy Complications. Circulation 2019, 139, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Kramer, C.K.; Campbell, S.; Retnakaran, R. Gestational diabetes and the risk of cardiovascular disease in women: A systematic review and meta-analysis. Diabetologia 2019, 62, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Shah, B.R.; Retnakaran, R.; Booth, G.L. Increased risk of cardiovascular disease in young women following gestational diabetes mellitus. Diabetes Care 2008, 31, 1668–1669. [Google Scholar] [CrossRef] [Green Version]
- Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; McIntyre, H.D.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [PubMed] [Green Version]
- Horvath, K.; Koch, K.; Jeitler, K.; Matyas, E.; Bender, R.; Bastian, H.; Lange, S.; Siebenhofer, A. Effects of treatment in women with gestational diabetes mellitus: Systematic review and meta-analysis. BMJ 2010, 340, c1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, T.A.; Xiang, A.H.; Page, K.A. Gestational diabetes mellitus: Risks and management during and after pregnancy. Nat. Rev. Endocrinol. 2012, 8, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, B.E.; Gabbe, S.G.; Persson, B.; Buchanan, T.A.; Catalano, P.A.; Damm, P.; Dyer, A.R.; Leiva, A.; Hod, M.; Kitzmiler, J.L.; et al. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care 2010, 33, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Kapur, A.; McIntyre, H.D.; Divakar, H.; Di Renzo, G.C.; Kihara, A.B.; McAuliffe, F.; Hanson, M.; Ma, R.C.; Hod, M.; PregFIGO Working Group on Hyperglycemia in Pregnancy. Towards a global consensus on GDM diagnosis: Light at the end of the tunnel? Int. J. Gynaecol. Obstet. 2020, 149, 257–261. [Google Scholar] [CrossRef]
- Feig, D.S.; Hwee, J.; Shah, B.R.; Booth, G.L.; Bierman, A.S.; Lipscombe, L.L. Trends in incidence of diabetes in pregnancy and serious perinatal outcomes: A large, population-based study in Ontario, Canada, 1996–2010. Diabetes Care 2014, 37, 1590–1596. [Google Scholar] [CrossRef] [Green Version]
- Franks, P.W.; Looker, H.C.; Kobes, S.; Touger, L.; Tataranni, P.A.; Hanson, R.L.; Knowler, W.C. Gestational glucose tolerance and risk of type 2 diabetes in young Pima Indian offspring. Diabetes 2006, 55, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Wicklow, B.A.; Sellers, E.A.C.; Sharma, A.K.; Kroeker, K.; Nickel, N.C.; Philips-Beck, W.; Shen, G.X. Association of Gestational Diabetes and Type 2 Diabetes Exposure In Utero With the Development of Type 2 Diabetes in First Nations and Non-First Nations Offspring. JAMA Pediatr. 2018, 172, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Sellers, E.A.; Dean, H.J.; Shafer, L.A.; Martens, P.J.; Phillips-Beck, W.; Heaman, M.; Prior, H.J.; Dart, A.B.; McGavock, J.; Morris, M.; et al. Exposure to Gestational Diabetes Mellitus: Impact on the Development of Early-Onset Type 2 Diabetes in Canadian First Nations and Non-First Nations Offspring. Diabetes Care 2016, 39, 2240–2246. [Google Scholar] [CrossRef] [Green Version]
- Clausen, T.D.; Mathiesen, E.R.; Hansen, T.; Pedersen, O.; Jensen, D.M.; Lauenborg, J.; Damm, P. High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes: The role of intrauterine hyperglycemia. Diabetes Care 2008, 31, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabelea, D.; Hanson, R.L.; Lindsay, R.S.; Pettitt, D.J.; Imperatore, G.; Gabir, M.M.; Roumain, J.; Bennett, P.H.; Knowler, W.C. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: A study of discordant sibships. Diabetes 2000, 49, 2208–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabelea, D. The predisposition to obesity and diabetes in offspring of diabetic mothers. Diabetes Care 2007, 30, S169–S174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabelea, D.; Mayer-Davis, E.J.; Lamichhane, A.P.; D’Agostino, R.B., Jr.; Liese, A.D.; Vehik, K.S.; Narayan, K.M.; Zeitler, P.; Hamman, R.F. Association of intrauterine exposure to maternal diabetes and obesity with type 2 diabetes in youth: The SEARCH Case-Control Study. Diabetes Care 2008, 31, 1422–1426. [Google Scholar] [CrossRef] [Green Version]
- Dabelea, D.; Stafford, J.M.; Mayer-Davis, E.J.; D’Agostino, R., Jr.; Dolan, L.; Imperatore, G.; Linder, B.; Lawrence, J.M.; Marcovina, S.M.; Mottl, A.K.; et al. Association of Type 1 Diabetes vs. Type 2 Diabetes Diagnosed During Childhood and Adolescence With Complications During Teenage Years and Young Adulthood. JAMA 2017, 317, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Turan, S.; Turan, O.M.; Miller, J.; Harman, C.; Reece, E.A.; Baschat, A.A. Decreased fetal cardiac performance in the first trimester correlates with hyperglycemia in pregestational maternal diabetes. Ultrasound Obs. Gynecol. 2011, 38, 325–331. [Google Scholar] [CrossRef]
- Wong, S.F.; Chan, F.Y.; Cincotta, R.B.; McIntyre, H.D.; Oats, J.J. Cardiac function in fetuses of poorly-controlled pre-gestational diabetic pregnancies—A pilot study. Gynecol. Obs. Investig. 2003, 56, 113–116. [Google Scholar] [CrossRef]
- Agoudemos, M.; Reinking, B.E.; Koppenhafer, S.L.; Segar, J.L.; Scholz, T.D. Programming of adult cardiovascular disease following exposure to late-gestation hyperglycemia. Neonatology 2011, 100, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Aman, J.; Hansson, U.; Ostlund, I.; Wall, K.; Persson, B. Increased fat mass and cardiac septal hypertrophy in newborn infants of mothers with well-controlled diabetes during pregnancy. Neonatology 2011, 100, 147–154. [Google Scholar] [CrossRef]
- Sheehan, P.Q.; Rowland, T.W.; Shah, B.L.; McGravey, V.J.; Reiter, E.O. Maternal diabetic control and hypertrophic cardiomyopathy in infants of diabetic mothers. Clin. Pediatr. 1986, 25, 266–271. [Google Scholar] [CrossRef]
- Clausen, T.D.; Mathiesen, E.R.; Hansen, T.; Pedersen, O.; Jensen, D.M.; Lauenborg, J.; Schmidt, L.; Damm, P. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 2464–2470. [Google Scholar] [CrossRef] [Green Version]
- Catalano, P.M.; McIntyre, H.D.; Cruickshank, J.K.; McCance, D.R.; Dyer, A.R.; Metzger, B.E.; Lowe, L.P.; Trimble, E.R.; Coustan, D.R.; Hadden, D.R.; et al. The hyperglycemia and adverse pregnancy outcome study: Associations of GDM and obesity with pregnancy outcomes. Diabetes Care 2012, 35, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Guillemette, L.; Wicklow, B.; Sellers, E.A.C.; Dart, A.; Shen, G.X.; Dolinsky, V.W.; Gordon, J.W.; Jassal, D.S.; Nickel, N.; Duhamel, T.A.; et al. Intrauterine exposure to diabetes and risk of cardiovascular disease in adolescence and early adulthood: A population-based birth cohort study. CMAJ 2020, 192, E1104–E1113. [Google Scholar] [CrossRef] [PubMed]
- Do, V.; Eckersley, L.; Lin, L.; Davidge, S.T.; Strickland, M.K.; Ojala, T.; Serrano-Lomelin, J.; Hornberger, L.K. Persistent Aortic Stiffness and Left Ventricular Hypertrophy in Children of Diabetic Mothers. Can. J. Cardiol. Open 2021, 3, 345–353. [Google Scholar] [CrossRef]
- Do, V.; Al-Hashmi, H.; Ojala, T.; Jain, V.; Colen, T.; Goncalvez-Alvarez, S.; Davidge, S.T.; Al-Rajaa, N.; Serrano-Lomelin, J.; Stickland, M.K.; et al. Cardiovascular Health of Offspring of Diabetic Mothers From the Fetal Through Late-Infancy Stages. JACC Cardiovasc. Imaging 2019, 12, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Pasek, R.C.; Gannon, M. Advancements and challenges in generating accurate animal models of gestational diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1327–E1338. [Google Scholar] [CrossRef] [Green Version]
- Rueda-Clausen, C.F.; Morton, J.S.; Davidge, S.T. The early origins of cardiovascular health and disease: Who, when, and how. Semin. Reprod. Med. 2011, 29, 197–210. [Google Scholar] [CrossRef]
- Aerts, L.; Vercruysse, L.; Van Assche, F.A. The endocrine pancreas in virgin and pregnant offspring of diabetic pregnant rats. Diabetes Res. Clin. Pract. 1997, 38, 9–19. [Google Scholar] [CrossRef]
- Holemans, K.; Aerts, L.; Van Assche, F.A. Evidence for an insulin resistance in the adult offspring of pregnant streptozotocin-diabetic rats. Diabetologia 1991, 34, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Xu, J.; Long, Y.S.; Epstein, P.N.; Liu, Y.Q. Rat maternal diabetes impairs pancreatic beta-cell function in the offspring. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E228–E236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Twinn, D.S.; Blackmore, H.L.; Siggens, L.; Giussani, D.A.; Cross, C.M.; Foo, R.; Ozanne, S.E. The programming of cardiac hypertrophy in the offspring by maternal obesity is associated with hyperinsulinemia, AKT, ERK, and mTOR activation. Endocrinology 2012, 153, 5961–5971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelsson, A.M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C.; et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oben, J.A.; Mouralidarane, A.; Samuelsson, A.M.; Matthews, P.J.; Morgan, M.L.; McKee, C.; Soeda, J.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Ozanne, S.E.; et al. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J. Hepatol. 2010, 52, 913–920. [Google Scholar] [CrossRef]
- Mouralidarane, A.; Soeda, J.; Visconti-Pugmire, C.; Samuelsson, A.M.; Pombo, J.; Maragkoudaki, X.; Butt, A.; Saraswati, R.; Novelli, M.; Fusai, G.; et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013, 58, 128–138. [Google Scholar] [CrossRef]
- Khan, I.Y.; Dekou, V.; Douglas, G.; Jensen, R.; Hanson, M.A.; Poston, L.; Taylor, P.D. A high-fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R127–R133. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Twinn, D.S.; Alfaradhi, M.Z.; Martin-Gronert, M.S.; Duque-Guimaraes, D.E.; Piekarz, A.; Ferland-McCollough, D.; Bushell, M.; Ozanne, S.E. Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol. Metab. 2014, 3, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Alfaradhi, M.Z.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Musial, B.; Fowden, A.; Ozanne, S.E. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R26–R34. [Google Scholar] [CrossRef]
- Khan, I.Y.; Taylor, P.D.; Dekou, V.; Seed, P.T.; Lakasing, L.; Graham, D.; Dominiczak, A.F.; Hanson, M.A.; Poston, L. Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension 2003, 41, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef]
- Pereira, T.J.; Fonseca, M.A.; Campbell, K.E.; Moyce, B.L.; Cole, L.K.; Hatch, G.M.; Doucette, C.A.; Klein, J.; Aliani, M.; Dolinsky, V.W. Maternal obesity characterized by gestational diabetes increases the susceptibility of rat offspring to hepatic steatosis via a disrupted liver metabolome. J. Physiol. 2015, 593, 3181–3197. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Brar, N.; Morriseau, T.S.; Kereliuk, S.M.; Fonseca, M.A.; Cole, L.K.; Jha, A.; Xiang, B.; Hunt, K.L.; Seshadri, N.; et al. Gestational Diabetes Adversely Affects Pancreatic Islet Architecture and Function in the Male Rat Offspring. Endocrinology 2019, 160, 1907–1925. [Google Scholar] [CrossRef]
- Brawerman, G.M.; Kereliuk, S.M.; Brar, N.; Cole, L.K.; Seshadri, N.; Pereira, T.J.; Xiang, B.; Hunt, K.L.; Fonseca, M.A.; Hatch, G.M.; et al. Maternal resveratrol administration protects against gestational diabetes-induced glucose intolerance and islet dysfunction in the rat offspring. J. Physiol. 2019, 597, 4175–4192. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Rosa, S.C.; Martens, M.D.; Field, J.T.; Nguyen, L.; Kereliuk, S.M.; Hai, Y.; Chapman, D.; Diehl-Jones, W.; Aliani, M.; West, A.R.; et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 2021, 17, 2257–2272. [Google Scholar] [CrossRef] [PubMed]
- Mughal, W.; Nguyen, L.; Pustylnik, S.; da Silva Rosa, S.C.; Piotrowski, S.; Chapman, D.; Du, M.; Alli, N.S.; Grigull, J.; Halayko, A.J.; et al. A conserved MADS-box phosphorylation motif regulates differentiation and mitochondrial function in skeletal, cardiac, and smooth muscle cells. Cell Death Dis. 2015, 6, e1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Pereira, T.J.; Moyce, B.L.; Mahood, T.H.; Doucette, C.A.; Rempel, J.; Dolinsky, V.W. In utero exposure to gestational diabetes mellitus conditions TLR4 and TLR2 activated IL-1beta responses in spleen cells from rat offspring. Biochim. Biophys. Acta 2016, 1862, 2137–2146. [Google Scholar] [CrossRef]
- Vuong, B.; Odero, G.; Rozbacher, S.; Stevenson, M.; Kereliuk, S.M.; Pereira, T.J.; Dolinsky, V.W.; Kauppinen, T.M. Exposure to gestational diabetes mellitus induces neuroinflammation, derangement of hippocampal neurons, and cognitive changes in rat offspring. J. Neuroinflamm. 2017, 14, 80. [Google Scholar] [CrossRef] [Green Version]
- Mdaki, K.S.; Larsen, T.D.; Wachal, A.L.; Schimelpfenig, M.D.; Weaver, L.J.; Dooyema, S.D.; Louwagie, E.J.; Baack, M.L. Maternal high-fat diet impairs cardiac function in offspring of diabetic pregnancy through metabolic stress and mitochondrial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H681–H692. [Google Scholar] [CrossRef] [Green Version]
- Louwagie, E.J.; Larsen, T.D.; Wachal, A.L.; Gandy, T.C.T.; Eclov, J.A.; Rideout, T.C.; Kern, K.A.; Cain, J.T.; Anderson, R.H.; Mdaki, K.S.; et al. Age and Sex Influence Mitochondria and Cardiac Health in Offspring Exposed to Maternal Glucolipotoxicity. iScience 2020, 23, 101746. [Google Scholar] [CrossRef]
- Blackmore, H.L.; Niu, Y.; Fernandez-Twinn, D.S.; Tarry-Adkins, J.L.; Giussani, D.A.; Ozanne, S.E. Maternal diet-induced obesity programs cardiovascular dysfunction in adult male mouse offspring independent of current body weight. Endocrinology 2014, 155, 3970–3980. [Google Scholar] [CrossRef] [Green Version]
- Turdi, S.; Ge, W.; Hu, N.; Bradley, K.M.; Wang, X.; Ren, J. Interaction between maternal and postnatal high fat diet leads to a greater risk of myocardial dysfunction in offspring via enhanced lipotoxicity, IRS-1 serine phosphorylation and mitochondrial defects. J. Mol. Cell Cardiol. 2013, 55, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Loche, E.; Blackmore, H.L.; Carpenter, A.A.; Beeson, J.H.; Pinnock, A.; Ashmore, T.J.; Aiken, C.E.; de Almeida-Faria, J.; Schoonejans, J.M.; Giussani, D.A.; et al. Maternal diet-induced obesity programmes cardiac dysfunction in male mice independently of post-weaning diet. Cardiovasc. Res. 2018, 114, 1372–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleao, L.C.; Inzani, I.; Furse, S.; Loche, E.; Hufnagel, A.; Ashmore, T.; Blackmore, H.L.; Jenkins, B.; Carpenter, A.A.M.; Wilczynska, A.; et al. Maternal diet-induced obesity during pregnancy alters lipid supply to mouse E18.5 fetuses and changes the cardiac tissue lipidome in a sex-dependent manner. Elife 2022, 11, e69078. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.D.; Sabey, K.H.; Knutson, A.J.; Gandy, T.C.T.; Louwagie, E.J.; Lauterboeck, L.; Mdaki, K.S.; Baack, M.L. Diabetic Pregnancy and Maternal High-Fat Diet Impair Mitochondrial Dynamism in the Developing Fetal Rat Heart by Sex-Specific Mechanisms. Int. J. Mol. Sci. 2019, 20, 3090. [Google Scholar] [CrossRef] [Green Version]
- Nakano, H.; Minami, I.; Braas, D.; Pappoe, H.; Wu, X.; Sagadevan, A.; Vergnes, L.; Fu, K.; Morselli, M.; Dunham, C.; et al. Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. Elife 2017, 6, e29330. [Google Scholar] [CrossRef]
- Lehtoranta, L.; Koskinen, A.; Vuolteenaho, O.; Laine, J.; Kyto, V.; Soukka, H.; Ekholm, E.; Rasanen, J. Gestational hyperglycemia reprograms cardiac gene expression in rat offspring. Pediatr. Res. 2017, 82, 356–361. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Gong, L.; Zhang, P.; Li, Y.; Liu, B.; Zhang, L.; Zhuang, J.; Xiao, D. Epigenetic Down-Regulation of Sirt 1 via DNA Methylation and Oxidative Stress Signaling Contributes to the Gestational Diabetes Mellitus-Induced Fetal Programming of Heart Ischemia-Sensitive Phenotype in Late Life. Int. J. Biol. Sci. 2019, 15, 1240–1251. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Wu, Y.; Lu, X.; Chidiac, P.; Wang, G.; Feng, Q. Maternal diabetes up-regulates NOX2 and enhances myocardial ischaemia/reperfusion injury in adult offspring. J. Cell Mol. Med. 2018, 22, 2200–2209. [Google Scholar] [CrossRef] [Green Version]
- Su, D.; Li, Y.; Guan, L.; Li, Q.; Shi, C.; Ma, X.; Song, Y. Elevated MST1 leads to apoptosis via depletion of YAP1 in cardiomyocytes exposed to high glucose. Mol. Med. 2021, 27, 13. [Google Scholar] [CrossRef]
- Preston, C.C.; Larsen, T.D.; Eclov, J.A.; Louwagie, E.J.; Gandy, T.C.T.; Faustino, R.S.; Baack, M.L. Maternal High Fat Diet and Diabetes Disrupts Transcriptomic Pathways That Regulate Cardiac Metabolism and Cell Fate in Newborn Rat Hearts. Front. Endocrinol. 2020, 11, 570846. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, B.; Larsen, T.; Barwari, S.; Louwagie, E.J.; Baack, M.L.; Dey, M. Prenatal Exposure to a Maternal High-Fat Diet Affects Histone Modification of Cardiometabolic Genes in Newborn Rats. Nutrients 2017, 9, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitmann, S.; Matrai, P.; Nemeth, D.; Hegyi, P.; Lukacs, A.; Berczi, B.; Czumbel, L.M.; Kiss, I.; Gyongyi, Z.; Varga, G.; et al. Maternal overnutrition elevates offspring’s blood pressure-A systematic review and meta-analysis. Paediatr. Perinat. Epidemiol. 2022, 36, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.; Vickers, M.H.; Segovia, S.A.; Zhang, X.D.; Reynolds, C.M. A maternal high fat diet programmes endothelial function and cardiovascular status in adult male offspring independent of body weight, which is reversed by maternal conjugated linoleic acid (CLA) supplementation. PLoS ONE 2015, 10, e0115994. [Google Scholar]
- Payen, C.; Guillot, A.; Paillat, L.; Fothi, A.; Dib, A.; Bourreau, J.; Schmitt, F.; Loufrani, L.; Aranyi, T.; Henrion, D.; et al. Pathophysiological adaptations of resistance arteries in rat offspring exposed in utero to maternal obesity is associated with sex-specific epigenetic alterations. Int. J. Obes. 2021, 45, 1074–1085. [Google Scholar] [CrossRef]
- Fan, L.; Lindsley, S.R.; Comstock, S.M.; Takahashi, D.L.; Evans, A.E.; He, G.W.; Thornburg, K.L.; Grove, K.L. Maternal high-fat diet impacts endothelial function in nonhuman primate offspring. Int. J. Obes. 2013, 37, 254–262. [Google Scholar] [CrossRef] [Green Version]
- Samuelsson, A.M.; Morris, A.; Igosheva, N.; Kirk, S.L.; Pombo, J.M.; Coen, C.W.; Poston, L.; Taylor, P.D. Evidence for sympathetic origins of hypertension in juvenile offspring of obese rats. Hypertension 2010, 55, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Armitage, J.A.; Lakasing, L.; Taylor, P.D.; Balachandran, A.A.; Jensen, R.I.; Dekou, V.; Ashton, N.; Nyengaard, J.R.; Poston, L. Developmental programming of aortic and renal structure in offspring of rats fed fat-rich diets in pregnancy. J. Physiol. 2005, 565, 171–184. [Google Scholar] [CrossRef]
- Carter, L.G.; Qi, N.R.; De Cabo, R.; Pearson, K.J. Maternal exercise improves insulin sensitivity in mature rat offspring. Med. Sci. Sports Exerc. 2013, 45, 832–840. [Google Scholar] [CrossRef] [Green Version]
- Carter, L.G.; Lewis, K.N.; Wilkerson, D.C.; Tobia, C.M.; Ngo Tenlep, S.Y.; Shridas, P.; Garcia-Cazarin, M.L.; Wolff, G.; Andrade, F.H.; Charnigo, R.J.; et al. Perinatal exercise improves glucose homeostasis in adult offspring. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1061–E1068. [Google Scholar] [CrossRef] [Green Version]
- Quiclet, C.; Dubouchaud, H.; Berthon, P.; Sanchez, H.; Vial, G.; Siti, F.; Fontaine, E.; Batandier, C.; Couturier, K. Maternal exercise modifies body composition and energy substrates handling in male offspring fed a high-fat/high-sucrose diet. J. Physiol. 2017, 595, 7049–7062. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Lee, M.Y.; Getchell, K.M.; So, K.; Hirshman, M.F.; Goodyear, L.J. Exercise before and during pregnancy prevents the deleterious effects of maternal high-fat feeding on metabolic health of male offspring. Diabetes 2015, 64, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, K.I.; Takahashi, H.; So, K.; Alves-Wagner, A.B.; Prince, N.B.; Lehnig, A.C.; Getchell, K.M.; Lee, M.Y.; Hirshman, M.F.; Goodyear, L.J. Maternal Exercise Improves Glucose Tolerance in Female Offspring. Diabetes 2017, 66, 2124–2136. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Twinn, D.S.; Gascoin, G.; Musial, B.; Carr, S.; Duque-Guimaraes, D.; Blackmore, H.L.; Alfaradhi, M.Z.; Loche, E.; Sferruzzi-Perri, A.N.; Fowden, A.L.; et al. Exercise rescues obese mothers’ insulin sensitivity, placental hypoxia and male offspring insulin sensitivity. Sci. Rep. 2017, 7, 44650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeson, J.H.; Blackmore, H.L.; Carr, S.K.; Dearden, L.; Duque-Guimaraes, D.E.; Kusinski, L.C.; Pantaleao, L.C.; Pinnock, A.G.; Aiken, C.E.; Giussani, D.A.; et al. Maternal exercise intervention in obese pregnancy improves the cardiovascular health of the adult male offspring. Mol. Metab. 2018, 16, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Boonpattrawong, N.P.; Golbidi, S.; Tai, D.C.; Aleliunas, R.E.; Bernatchez, P.; Miller, J.W.; Laher, I.; Devlin, A.M. Exercise during pregnancy mitigates the adverse effects of maternal obesity on adult male offspring vascular function and alters one-carbon metabolism. Physiol. Rep. 2020, 8, e14582. [Google Scholar] [CrossRef] [PubMed]
- Saiyin, T.; Engineer, A.; Greco, E.R.; Kim, M.Y.; Lu, X.; Jones, D.L.; Feng, Q. Maternal voluntary exercise mitigates oxidative stress and incidence of congenital heart defects in pre-gestational diabetes. J. Cell Mol. Med. 2019, 23, 5553–5565. [Google Scholar] [CrossRef]
- Guillemette, L.; Hay, J.L.; Kehler, D.S.; Hamm, N.C.; Oldfield, C.; McGavock, J.M.; Duhamel, T.A. Exercise in Pregnancy and Children’s Cardiometabolic Risk Factors: A Systematic Review and Meta-Analysis. Sports Med. Open 2018, 4, 35. [Google Scholar] [CrossRef] [Green Version]
- Brawerman, G.M.; Dolinsky, V.W. Therapies for gestational diabetes and their implications for maternal and offspring health: Evidence from human and animal studies. Pharm. Res. 2018, 130, 52–73. [Google Scholar] [CrossRef]
- Cole, L.K.; Zhang, M.; Chen, L.; Sparagna, G.C.; Vandel, M.; Xiang, B.; Dolinsky, V.W.; Hatch, G.M. Supplemental Berberine in a High-Fat Diet Reduces Adiposity and Cardiac Dysfunction in Offspring of Mouse Dams with Gestational Diabetes Mellitus. J. Nutr. 2021, 151, 892–901. [Google Scholar] [CrossRef]
- Cole, L.K.; Sparagna, G.C.; Vandel, M.; Xiang, B.; Dolinsky, V.W.; Hatch, G.M. Berberine elevates cardiolipin in heart of offspring from mouse dams with high fat diet-induced gestational diabetes mellitus. Sci. Rep. 2021, 11, 15770. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; He, B.; Chen, Z.; Yan, M.; Wu, L. Anti-inflammatory activity of berberine in non-alcoholic fatty liver disease via the Angptl2 pathway. BMC Immunol. 2020, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Baggioni, A. Berberine and Its Role in Chronic Disease. Adv. Exp. Med. Biol. 2016, 928, 27–45. [Google Scholar] [PubMed]
- Yeung, A.W.K.; Orhan, I.E.; Aggarwal, B.B.; Battino, M.; Belwal, T.; Bishayee, A.; Daglia, M.; Georgieva, M.G.; Gupta, V.K.; Horbanczuk, J.O.; et al. Berberine, a popular dietary supplement for human and animal health: Quantitative research literature analysis—A review. Anim. Sci. Pap. Rep. 2020, 38, 5–19. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kereliuk, S.M.; Dolinsky, V.W. Recent Experimental Studies of Maternal Obesity, Diabetes during Pregnancy and the Developmental Origins of Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 4467. https://doi.org/10.3390/ijms23084467
Kereliuk SM, Dolinsky VW. Recent Experimental Studies of Maternal Obesity, Diabetes during Pregnancy and the Developmental Origins of Cardiovascular Disease. International Journal of Molecular Sciences. 2022; 23(8):4467. https://doi.org/10.3390/ijms23084467
Chicago/Turabian StyleKereliuk, Stephanie M., and Vernon W. Dolinsky. 2022. "Recent Experimental Studies of Maternal Obesity, Diabetes during Pregnancy and the Developmental Origins of Cardiovascular Disease" International Journal of Molecular Sciences 23, no. 8: 4467. https://doi.org/10.3390/ijms23084467