
Inhibiting Endocannabinoid Hydrolysis as Emerging Analgesic Strategy Targeting a Spectrum of Ion Channels Implicated in Migraine Pain


Abstract
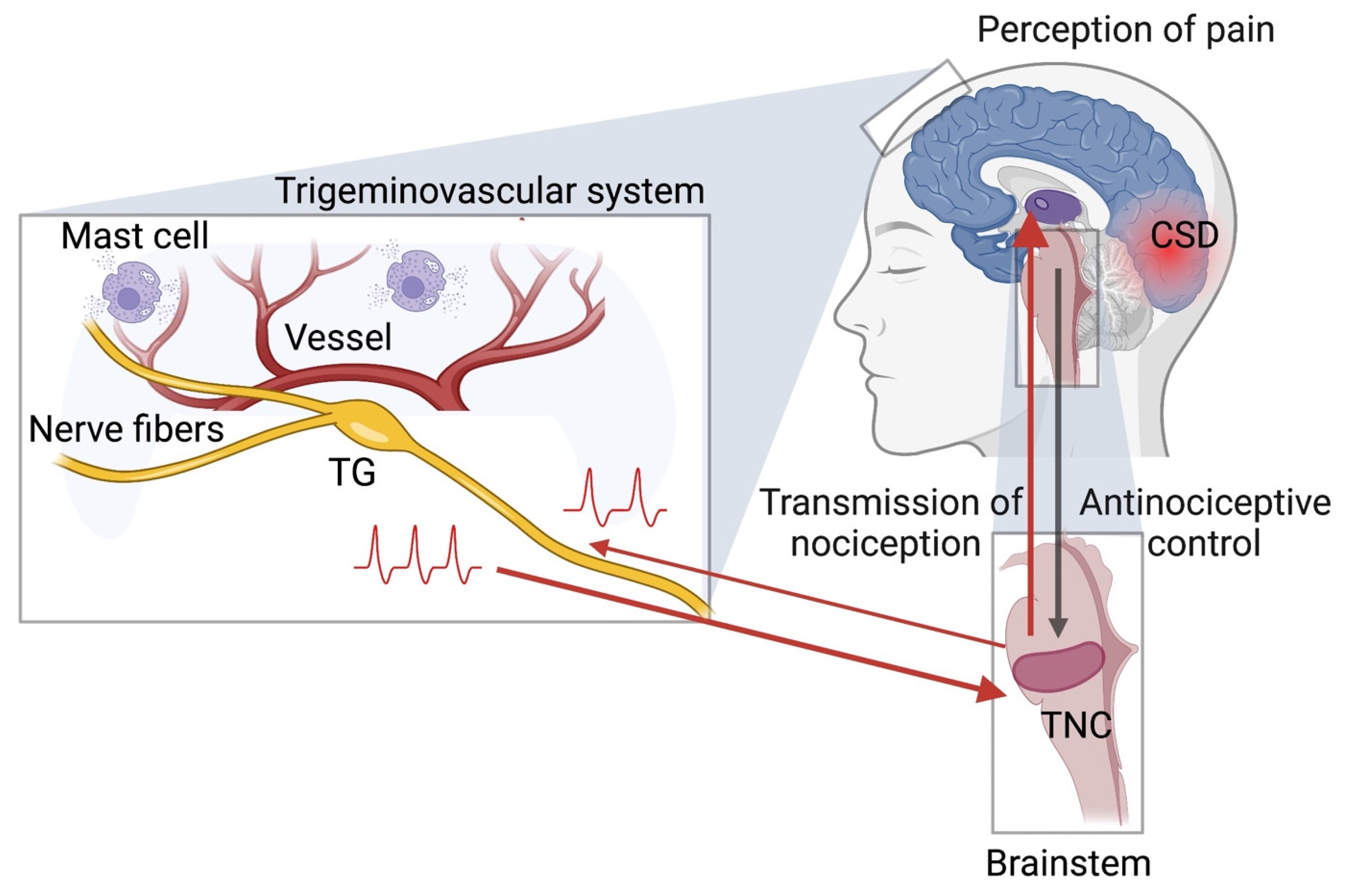
:1. Introduction: Migraine Pain Signaling Pathways as Target for Antinociception
2. ECS in Anatomical Structures Important for Migraine Pain Signaling
2.1. Main Components of the ECS as a Target for Analgesia
2.2. MAGL and FAAH Activity in Migraine-Related Areas of the Nervous System
3. EndoCBs Control of Nociception via Cannabinoid Receptors and through the Direct Action on Ion Channels
3.1. Distribution of CB1 and CB2 Receptors and Retrograde endoCB Signaling in the Nociceptive System
3.2. Pro-Nociceptive Effects of EndoCBs via TRPV1 Receptors
3.3. Modulation of Nociception by EndoCBs via Membrane Lipid Environment and Direct Interaction with Ion Channels
4. MAGL and FAAH Inhibition to Treat Migraine Pain
4.1. Current Approaches to Treat Migraine Pain and the Need for New Treatment Options
4.2. Preventing Endocannabinoid Hydrolysis as a Novel Analgesic Strategy
4.3. ECS as a Target for Treating Migraine with Aura?

{kind=link}
{kind=link}
{kind=link}
| Inhibitors | Compounds | IC50 | Analgesic Effects and Targets | Ref |
|---|---|---|---|---|
| FAAH |  OL135 | 5 nM | Attenuation of mechanical and cold allodynia | [112] |
 PF3845 | 514 nM | Attenuation of mechanical and cold allodynia | [119,120,151] | |
 URB597 | 5 nM | Moderate thermal antinociception. Anti-allodynic effect in inflammatory pain. Decreased hyperalgesia in the TGVS | [115,116,122] | |
 URB937 | 26.8 nM | Inhibition of nocifensive behavior. Decreased peripheral nociception | [121,152] | |
| MAGL |  JZL184 | 262 nM | Behavioral analgesic effects. Reduction of NTG-induced hyperalgesia of spinal and TGVS origin | [130,131,135,136] |
 URB602 | 280 nM | Reduction of NTG-induced hyperalgesia of spinal and TGVS origin | [129,131] | |
 KML29 | 43 nm | Behavioral analgesic effect | [45,132,133,134] | |
 MJN110 | <100 nM | Attenuation of mechanical allodynia and thermal hyperalgesia | [138,139] | |
 JJKK-048 | <0.4 nM | Not tested | [123] | |
| Dual MAGL FAAH |  JZL195 | 13 nM FAAH 19 nM MAGL | Reduction of peripheral and cephalic pain | [145] |
 AKU-005 | 0.2–1.1 nM MAGL 63 nM FAAH | Not tested | [141] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoyl glycerol |
| ACC | Anterior cingulate cortex |
| AEA | N-arachidonoyl ethanolamide, anandamide |
| BoNT-A | Botulinum neurotoxin serotype A |
| CB1/2 | Cannabinoid receptors 1, 2 |
| CGRP | Calcitonin gene related peptide |
| CNS | Central nervous system |
| CSD | Cortical spreading depression |
| DAGL | Diacylglycerol lipase |
| ECS | Endocannabinoid system |
| EndoCBs | Endocannabinoids |
| FAAH | Fatty acid amide hydrolase |
| MAGL | Monoacylglycerol lipase |
| mGluR | Metabotropic glutamate receptor |
| NAPE-PLD | NAPE-specific phospholipase D |
| NMDAR | N-methyl-D-aspartate receptors |
| PNS | Peripheral nervous system |
| TG | Trigeminal ganglion |
| TGVS | Trigeminovascular system |
| TRPM3 | Transient Receptor Potential Cation Channel Subfamily M Member 3 |
| TRPV1 | Transient receptor potential vanilloid 1 |
| VGCC | Voltage-gated calcium channel |
References
- Vincent, M.; Wang, S. Headache classification committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia 2018, 38, 1–211. [Google Scholar] [CrossRef]
- Schaible, H. Peripheral and Central Mechanisms of pain generation. In Handbook of Experimental Pharmacology; 2006; Volume 177, pp. 3–28. [Google Scholar]
- Meacham, K.; Shepherd, A.; Mohapatra, D.P.; Haroutounian, S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr. Pain Headache Rep. 2017, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Romero, J.; Reinhard, J.F.; Melamed, E.; Pettibone, D.J. Neurotransmitters and the fifth cranial nerve: Is there a relation to the headache phase of migraine? Lancet 1979, 314, 883–885. [Google Scholar] [CrossRef]
- Ashina, M.; Hansen, J.M.; Do, T.P.; Melo-Carrillo, A.; Burstein, R.; Moskowitz, M.A. Migraine and the trigeminovascular system—40 years and counting. Lancet Neurol. 2019, 4422, 795–804. [Google Scholar] [CrossRef]
- Levy, D. Migraine pain, meningeal inflammation, and mast cells. Curr. Pain Headache Rep. 2009, 13, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Noseda, R.; Burstein, R. Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain 2013, 154, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Koroleva, K.; Gafurov, O.; Guselnikova, V.; Nurkhametova, D.; Giniatullina, R.; Sitdikova, G.; Mattila, O.S.; Lindsberg, P.J.; Malm, T.M.; Giniatullin, R. Meningeal Mast Cells Contribute to ATP-Induced Nociceptive Firing in Trigeminal Nerve Terminals: Direct and Indirect Purinergic Mechanisms Triggering Migraine Pain. Front. Cell. Neurosci. 2019, 13, 195. [Google Scholar] [CrossRef]
- Shelukhina, I.; Mikhailov, N.; Abushik, P.; Nurullin, L.; Nikolsky, E.E.; Giniatullin, R. Cholinergic nociceptive mechanisms in rat meninges and trigeminal Ganglia: Potential implications for migraine pain. Front. Neurol. 2017, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Giniatullin, R. 5-hydroxytryptamine in migraine: The puzzling role of ionotropic 5-HT 3 receptor in the context of established therapeutic effect of metabotropic 5-HT 1 subtypes. Br. J. Pharmacol. 2022, 179, 400–415. [Google Scholar] [CrossRef]
- Della Pietra, A.; Mikhailov, N.; Giniatullin, R. The emerging role of mechanosensitive piezo channels in migraine pain. Int. J. Mol. Sci. 2020, 21, 696. [Google Scholar] [CrossRef] [Green Version]
- Gafurov, O.; Koroleva, K.; Giniatullin, R. Antidromic Spike Propagation and Dissimilar Expression of P2X, 5-HT, and TRPV1 Channels in Peripheral vs. Central Sensory Axons in Meninges. Front. Cell. Neurosci. 2021, 14, 471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-C.; Strassman, A.M.; Burstein, R.; Levy, D. Sensitization and activation of intracranial meningeal nociceptors by mast cell mediators. J. Pharmacol. Exp. Ther. 2007, 322, 806–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strassman, A.M.; Raymond, S.A.; Burstein, R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature 1996, 384, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Karatas, H.; Erdener, S.E.; Gursoy-Ozdemir, Y.; Lule, S.; Eren-Koçak, E.; Sen, Z.D.; Dalkara, T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013, 339, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Messlinger, K. The big CGRP flood-sources, sinks and signalling sites in the trigeminovascular system. J. Headache Pain 2018, 19, 22. [Google Scholar] [CrossRef] [Green Version]
- Messlinger, K.; Russo, A.F. Current understanding of trigeminal ganglion structure and function in headache. Cephalalgia 2019, 39, 1661–1674. [Google Scholar] [CrossRef]
- Edvinsson, J.C.A.; Maddahi, A.; Christiansen, I.M.; Reducha, P.V.; Warfvinge, K.; Sheykhzade, M.; Edvinsson, L.; Haanes, K.A. Lasmiditan and 5-Hydroxytryptamine in the rat trigeminal system; expression, release and interactions with 5-HT1 receptors. J. Headache Pain 2022, 23, 26. [Google Scholar] [CrossRef]
- Apkarian, A.V.; Reckziegel, D. Peripheral and central viewpoints of chronic pain, and translational implications. Neurosci. Lett. 2019, 702, 3–5. [Google Scholar] [CrossRef]
- Vachon-Presseau, E.; Tétreault, P.; Petre, B.; Huang, L.; Berger, S.E.; Torbey, S.; Baria, A.T.; Mansour, A.R.; Hashmi, J.A.; Griffith, J.W.; et al. Corticolimbic anatomical characteristics predetermine risk for chronic pain. Brain 2016, 139, 1958–1970. [Google Scholar] [CrossRef] [Green Version]
- Bolay, H.; Vuralli, D.; Goadsby, P.J. Aura and Head pain: Relationship and gaps in the translational models. J. Headache Pain 2019, 20, 94. [Google Scholar] [CrossRef] [Green Version]
- Tottene, A.; Conti, R.; Fabbro, A.; Vecchia, D.; Shapovalova, M.; Santello, M.; van den Maagdenberg, A.M.J.M.; Ferrari, M.D.; Pietrobon, D. Enhanced Excitatory Transmission at Cortical Synapses as the Basis for Facilitated Spreading Depression in CaV2.1 Knockin Migraine Mice. Neuron 2009, 61, 762–773. [Google Scholar] [CrossRef] [PubMed]
- van den Maagdenberg, A.M.J.; Pietrobon, D.; Pizzorusso, T.; Kaja, S.; Broos, L.A.; Cesetti, T.; van de Ven, R.C.; Tottene, A.; van der Kaa, J.; Plomp, J.J.; et al. A Cacna1a Knockin Migraine Mouse Model with Increased Susceptibility to Cortical Spreading Depression. Neuron 2004, 41, 701–710. [Google Scholar] [CrossRef]
- Burstein, R.; Yarnitsky, D.; Goor-Aryeh, I.; Ransil, B.J.; Bajwa, Z.H. An association between migraine and cutaneous allodynia. Ann. Neurol. 2000, 47, 614–624. [Google Scholar] [CrossRef]
- Mungoven, T.J.; Henderson, L.A.; Meylakh, N. Chronic Migraine Pathophysiology and Treatment: A Review of Current Perspectives. Front. Pain Res. 2021, 2, 52. [Google Scholar] [CrossRef] [PubMed]
- Benemei, S.; Bentivegna, E.; Martelletti, P. Positioning the new drugs for migraine. Expert Opin. Drug Metab. Toxicol. 2022, 18. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-F. Potassium channels as molecular targets of endocannabinoids. Channels 2021, 15, 408–423. [Google Scholar] [CrossRef]
- Savinainen, J.R.; Saario, S.M.; Laitinen, J.T. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol. 2012, 204, 267–276. [Google Scholar] [CrossRef]
- Hillard, C.J. The Endocannabinoid Signaling System in the CNS: A Primer. Int. Rev. Neurobiol. 2015, 125, 1–47. [Google Scholar]
- Katona, I. Molecular Composition of the Endocannabinoid System at Glutamatergic Synapses. J. Neurosci. 2006, 26, 5628–5637. [Google Scholar] [CrossRef]
- Mátyás, F.; Urbán, G.M.; Watanabe, M.; Mackie, K.; Zimmer, A.; Freund, T.F.; Katona, I. Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology 2008, 54, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horváth, E.; Woodhams, S.G.; Nyilas, R.; Henstridge, C.M.; Kano, M.; Sakimura, K.; Watanabe, M.; Katona, I. Heterogeneous presynaptic distribution of monoacylglycerol lipase, a multipotent regulator of nociceptive circuits in the mouse spinal cord. Eur. J. Neurosci. 2014, 39, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickel, V.M.; Shobin, E.T.; Lane, D.A.; Mackie, K. Cannabinoid-1 receptors in the mouse ventral pallidum are targeted to axonal profiles expressing functionally opposed opioid peptides and contacting N-acylphosphatidylethanolamine-hydrolyzing phospholipase D terminals. Neuroscience 2012, 227, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, N.; Yamanaka, K.; Yamamoto, S. Purification and Characterization of an Acid Amidase Selective for N-Palmitoylethanolamine, a Putative Endogenous Anti-inflammatory Substance. J. Biol. Chem. 2001, 276, 35552–35557. [Google Scholar] [CrossRef] [Green Version]
- Kaczocha, M.; Glaser, S.T.; Deutsch, D.G. Identification of intracellular carriers for the endocannabinoid anandamide. Proc. Natl. Acad. Sci. USA 2009, 106, 6375–6380. [Google Scholar] [CrossRef] [Green Version]
- Oddi, S.; Fezza, F.; Pasquariello, N.; D’Agostino, A.; Catanzaro, G.; De Simone, C.; Rapino, C.; Finazzi-Agrò, A.; Maccarrone, M. Molecular Identification of Albumin and Hsp70 as Cytosolic Anandamide-Binding Proteins. Chem. Biol. 2009, 16, 624–632. [Google Scholar] [CrossRef]
- Liedhegner, E.S.; Vogt, C.D.; Sem, D.S.; Cunningham, C.W.; Hillard, C.J. Sterol Carrier Protein-2: Binding Protein for Endocannabinoids. Mol. Neurobiol. 2014, 50, 149–158. [Google Scholar] [CrossRef] [Green Version]
- McFarland, M.J.; Terebova, E.A.; Barker, E.L. Detergent-resistant membrane microdomains in the disposition of the lipid signaling molecule anandamide. AAPS J. 2006, 8, E95–E100. [Google Scholar] [CrossRef] [Green Version]
- Chicca, A.; Marazzi, J.; Nicolussi, S.; Gertsch, J. Evidence for Bidirectional Endocannabinoid Transport across Cell Membranes. J. Biol. Chem. 2012, 287, 34660–34682. [Google Scholar] [CrossRef] [Green Version]
- Bisogno, T.; Maccarrone, M.; De Petrocellis, L.; Jarrahian, A.; Finazzi-Agrò, A.; Hillard, C.; Di Marzo, V. The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur. J. Biochem. 2001, 268, 1982–1989. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, Y.Q.; Chen, S.P.; Wang, X.M.; Xu, B.Y.; Li, D.Y.; Tian, Y.K.; Ye, D.W. The endocannabinoid system: Novel targets for treating cancer induced bone pain. Biomed. Pharmacother. 2019, 120, 109504. [Google Scholar] [CrossRef] [PubMed]
- Khademi, H.; Kamangar, F.; Brennan, P.; Malekzadeh, R. Opioid Therapy and its Side Effects: A Review. Arch. Iran. Med 2016, 19, 870–876. [Google Scholar] [PubMed]
- Elikottil, J.; Gupta, P.; Gupta, K. The analgesic potential of cannabinoids. J. Opioid Manag. 2009, 5, 341–357. [Google Scholar] [CrossRef]
- Ghosh, S.; Kinsey, S.G.; Liu, Q.-S.; Hruba, L.; McMahon, L.R.; Grim, T.W.; Merritt, C.R.; Wise, L.E.; Abdullah, R.A.; Selley, D.E.; et al. Full Fatty Acid Amide Hydrolase Inhibition Combined with Partial Monoacylglycerol Lipase Inhibition: Augmented and Sustained Antinociceptive Effects with Reduced Cannabimimetic Side Effects in Mice. J. Pharmacol. Exp. Ther. 2015, 354, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [Green Version]
- Della Pietra, A.; Giniatullin, R.; Savinainen, J.R. Distinct Activity of Endocannabinoid-Hydrolyzing Enzymes MAGL and FAAH in Key Regions of Peripheral and Central Nervous System Implicated in Migraine. Int. J. Mol. Sci. 2021, 22, 1204. [Google Scholar] [CrossRef]
- Sugiura, T.; Waku, K. Cannabinoid Receptors and Their Endogenous Ligands. J. Biochem. 2002, 132, 7–12. [Google Scholar] [CrossRef]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [Green Version]
- Cruz, S.L.; Sánchez-Miranda, E.; Castillo-Arellano, J.I.; Cervantes-Villagrana, R.D.; Ibarra-Sánchez, A.; González-Espinosa, C. Anandamide inhibits FcεRI-dependent degranulation and cytokine synthesis in mast cells through CB2 and GPR55 receptor activation. Possible involvement of CB2-GPR55 heteromers. Int. Immunopharmacol. 2018, 64, 298–307. [Google Scholar] [CrossRef]
- Levy, D. Endogenous Mechanisms Underlying the Activation and Sensitization of Meningeal Nociceptors: The Role of Immuno-Vascular Interactions and Cortical Spreading Depression. Curr. Pain Headache Rep. 2012, 16, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, E.; Ankarali, S.; Torun, I.E.; Dagistan, Y. Receptor mechanisms mediating the anti-neuroinflammatory effects of endocannabinoid system modulation in a rat model of migraine. Eur. J. Neurosci. 2022, 55, 1015–1031. [Google Scholar] [CrossRef] [PubMed]
- Leimuranta, P.; Khiroug, L.; Giniatullin, R. Emerging role of (endo)cannabinoids in migraine. Front. Pharmacol. 2018, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; Hohmann, A.G. The endocannabinoid system and pain. CNS Neurol. Disord. Drug Targets 2009, 8, 403–421. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.; Johnson, M.; Melvin, L.; de Costa, B.; Rice, K. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583. [Google Scholar] [CrossRef]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; De Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [Green Version]
- Cabral, G.A.; Marciano-Cabral, F. Cannabinoid receptors in microglia of the central nervous system: Immune functional relevance. J. Leukoc. Biol. 2005, 78, 1192–1197. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Neuroscience: Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Safiulina, V.F.; Kasyanov, A.M.; Giniatullin, R.; Cherubini, E. Adenosine down-regulates giant depolarizing potentials in the developing rat hippocampus by exerting a negative control on glutamatergic inputs. J. Neurophysiol. 2005, 94, 2797–2804. [Google Scholar] [CrossRef] [Green Version]
- Albayram, O.; Alferink, J.; Pitsch, J.; Piyanova, A.; Neitzert, K.; Poppensieker, K.; Mauer, D.; Michel, K.; Legler, A.; Becker, A.; et al. Role of CB1 cannabinoid receptors on GABAergic neurons in brain aging. Proc. Natl. Acad. Sci. USA 2011, 108, 11256–11261. [Google Scholar] [CrossRef] [Green Version]
- Azad, S.C.; Kurz, J.; Marsicano, G.; Lutz, B.; Zieglgänsberger, W.; Rammes, G. Activation of CB1 specifically located on GABAergic interneurons inhibits LTD in the lateral amygdala. Learn. Mem. 2008, 15, 143–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, A.F.; Laaris, N.; Kawamura, M.; Masino, S.A.; Lupica, C.R. Control of Cannabinoid CB1 Receptor Function on Glutamate Axon Terminals by Endogenous Adenosine Acting at A1 Receptors. J. Neurosci. 2010, 30, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Zorumski, C.F. Pharmacological Aspects of NMDA Receptors, mGluR5, and Endocannabinoids. In Handbook of Cannabis and Related Pathologies; Elsevier: Amsterdam, The Netherlands, 2017; pp. 630–638. [Google Scholar]
- Fischer, M.; Messlinger, K. Cannabinoid and Vanilloid Effects of R(+)-Methanandamide in the Hemisected Meningeal Preparation. Cephalalgia 2007, 27, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Lenz, R.A.; Alger, B.E. Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J. Physiol. 1999, 521, 147–157. [Google Scholar] [CrossRef]
- Diana, M.A.; Marty, A. Endocannabinoid-mediated short-term synaptic plasticity: Depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br. J. Pharmacol. 2004, 142, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Straiker, A.; Mackie, K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J. Physiol. 2005, 569, 501–517. [Google Scholar] [CrossRef]
- Iversen, L. Cannabinoids: A real prospect for pain relief. Curr. Opin. Pharmacol. 2002, 2, 50–55. [Google Scholar] [CrossRef]
- Pertwee, R.G. The pharmacology of cannabinoid receptors and their ligands: An overview. Int. J. Obes. 2006, 30, S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Piomelli, D.; Tassorelli, C. Endocannabinoid System and Migraine Pain: An Update. Front. Neurosci. 2018, 12, 172. [Google Scholar] [CrossRef]
- Alger, B.E.; Kim, J. Supply and demand for endocannabinoids. Trends Neurosci. 2011, 34, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.E.; Brown, S.; Hille, B.; Mackie, K. Protein Kinase C Disrupts Cannabinoid Actions by Phosphorylation of the CB1 Cannabinoid Receptor. J. Neurosci. 1998, 18, 2834–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladarre, D.; Roland, A.B.; Biedzinski, S.; Ricobaraza, A.; Lenkei, Z. Polarized cellular patterns of endocannabinoid production and detection shape cannabinoid signaling in neurons. Front. Cell. Neurosci. 2015, 8, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dux, M.; Deák, É.; Tassi, N.; Sántha, P.; Jancsó, G. Endovanilloids are potential activators of the trigeminovascular nocisensor complex. J. Headache Pain 2016, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Akerman, S.; Kaube, H.; Goadsby, P.J. Anandamide Is Able to Inhibit Trigeminal Neurons Using an in Vivo Model of Trigeminovascular-Mediated Nociception. J. Pharmacol. Exp. Ther. 2004, 309, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Zakharov, A.; Vitale, C.; Kilinc, E.; Koroleva, K.; Fayuk, D.; Shelukhina, I. Hunting for origins of migraine pain: Cluster analysis of spontaneous and capsaicin-induced firing in meningeal trigeminal nerve fibers. Front. Cell. Neurosci. 2015, 9, 287. [Google Scholar] [CrossRef]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef]
- Ryskamp, D.; Redmon, S.; Jo, A.; Križaj, D. TRPV1 and Endocannabinoids: Emerging Molecular Signals that Modulate Mammalian Vision. Cells 2014, 3, 914–938. [Google Scholar] [CrossRef]
- Tognetto, M.; Amadesi, S.; Harrison, S.; Creminon, C.; Trevisani, M.; Carreras, M.; Matera, M.; Geppetti, P.; Bianchi, A. Anandamide excites central terminals of dorsal root ganglion neurons via vanilloid receptor-1 activation. J. Neurosci. 2001, 21, 1104–1109. [Google Scholar] [CrossRef]
- Akopian, A.N.; Ruparel, N.B.; Jeske, N.A.; Patwardhan, A.; Hargreaves, K.M. Role of ionotropic cannabinoid receptors in peripheral antinociception and antihyperalgesi. trends Pharmacol. Sci. 2008, 30, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Suleimanova, A.; Talanov, M.; Gafurov, O.; Gafarov, F.; Koroleva, K.; Virenque, A.; Noe, F.M.; Mikhailov, N.; Nistri, A.; Giniatullin, R. Modeling a Nociceptive Neuro-Immune Synapse Activated by ATP and 5-HT in Meninges: Novel Clues on Transduction of Chemical Signals Into Persistent or Rhythmic Neuronal Firing. Front. Cell. Neurosci. 2020, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Suleimanova, A.; Talanov, M.; van den Maagdenberg, A.M.J.M.; Giniatullin, R. Deciphering in silico the Role of Mutated NaV1.1 Sodium Channels in Enhancing Trigeminal Nociception in Familial Hemiplegic Migraine Type 3. Front. Cell. Neurosci. 2021, 15, 202. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G.; Guerrero-Toro, C.; Kilinc, E.; Koroleva, K.; Ishchenko, Y.; Abushik, P.; Giniatullina, R.; Fayuk, D.; Giniatullin, R. Nucleotide homeostasis and purinergic nociceptive signaling in rat meninges in migraine-like conditions. Purinergic Signal. 2016, 12, 561–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailov, N.; Leskinen, J.; Fagerlund, I.; Poguzhelskaya, E.; Giniatullina, R.; Gafurov, O.; Malm, T.; Karjalainen, T.; Gröhn, O.; Giniatullin, R. Mechanosensitive meningeal nociception via Piezo channels: Implications for pulsatile pain in migraine? Neuropharmacology 2019, 149, 113–123. [Google Scholar] [CrossRef]
- Mikhailov, N.; Plotnikova, L.; Singh, P.; Giniatullin, R.; Hämäläinen, R.H. Functional Characterization of Mechanosensitive Piezo1 Channels in Trigeminal and Somatic Nerves in a Neuron-on-Chip Model. Int. J. Mol. Sci. 2022, 23, 1370. [Google Scholar] [CrossRef]
- Krivoshein, G.; Tolner, E.A.; van den Maagdenberg, A.; Giniatullin, R.A. Migraine-relevant sex-dependent activation of mouse meningeal afferents by TRPM3 agonists. J. Headache Pain 2022, 23, 4. [Google Scholar] [CrossRef]
- Borbiro, I.; Badheka, D.; Rohacs, T. Activation of TRPV1 channels inhibits mechanosensitive piezo channel activity by depleting membrane phosphoinositides. Sci. Signal. 2015, 8, ra15. [Google Scholar] [CrossRef] [Green Version]
- Romero, L.O.; Massey, A.E.; Mata-Daboin, A.D.; Sierra-Valdez, F.J.; Chauhan, S.C.; Cordero-Morales, J.F.; Vásquez, V. Dietary fatty acids fine-tune Piezo1 mechanical response. Nat. Commun. 2019, 10, 1200. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Honoré, E. Sensing pressure with ion channels. Trends Neurosci. 2012, 35, 477–486. [Google Scholar] [CrossRef]
- Carta, M.; Lanore, F.; Rebola, N.; Szabo, Z.; Da Silva, S.V.; Lourenço, J.; Verraes, A.; Nadler, A.; Schultz, C.; Blanchet, C.; et al. Membrane Lipids Tune Synaptic Transmission by Direct Modulation of Presynaptic Potassium Channels. Neuron 2014, 81, 787–799. [Google Scholar] [CrossRef] [Green Version]
- Ghovanloo, M.-R.; Ruben, P.C. Cannabidiol and Sodium Channel Pharmacology: General Overview, Mechanism, and Clinical Implications. Neurosci. 2021, 107385842110170. [Google Scholar] [CrossRef] [PubMed]
- Al Kury, L.T.; Voitychuk, O.I.; Yang, K.-H.S.; Thayyullathil, F.T.; Doroshenko, P.; Ramez, A.M.; Shuba, Y.M.; Galadari, S.; Howarth, F.C.; Oz, M. Effects of the endogenous cannabinoid anandamide on voltage-dependent sodium and calcium channels in rat ventricular myocytes. Br. J. Pharmacol. 2014, 171, 3485–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Liao, C.; Jain, S.; Nicholson, R.A. The cannabinoid receptor agonist CP-55,940 and ethyl arachidonate interfere with [3H] batrachotoxinin A 20 α-benzoate binding to sodium channels and inhibit sodium channel function. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2008, 148, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Imendra, K.G.; Miyazaki, T.; Hotokezaka, H.; Fujiyama, R.; Zeredo, J.L.; Miyamoto, T.; Toda, K. Biophysical properties of voltage-gated Na+ channels in frog parathyroid cells and their modulation by cannabinoids. J. Exp. Biol. 2005, 208, 4747–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eroli, F.; Loonen, I.C.M.; van den Maagdenberg, A.M.J.M.; Tolner, E.A.; Nistri, A. Differential neuromodulatory role of endocannabinoids in the rodent trigeminal sensory ganglion and cerebral cortex relevant to pain processing. Neuropharmacology 2018, 131, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Lipton, R.B.; Silberstein, S.D. Episodic and Chronic Migraine Headache: Breaking Down Barriers to Optimal Treatment and Prevention. Headache J. Head Face Pain 2015, 55, 103–122. [Google Scholar] [CrossRef]
- Weatherall, M.W. The diagnosis and treatment of chronic migraine. Ther. Adv. Chronic Dis. 2015, 6, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, M.; Goadsby, P.; Roon, K.; Lipton, R. Triptans (Serotonin, 5-HT 1B/1D Agonists) in Migraine: Detailed Results and Methods of A Meta-Analysis of 53 Trials. Cephalalgia 2002, 22, 633–658. [Google Scholar] [CrossRef]
- Linde, K.; Rossnagel, K. Propranolol for migraine prophylaxis. In Cochrane Database of Systematic Reviews; Hobson, A., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2004. [Google Scholar]
- Mulleners, W.M.; McCrory, D.C.; Linde, M. Antiepileptics in migraine prophylaxis: An updated Cochrane review. Cephalalgia 2015, 35, 51–62. [Google Scholar] [CrossRef]
- Diener, H.-C.; Bussone, G.; Van Oene, J.; Lahaye, M.; Schwalen, S.; Goadsby, P. Topiramate Reduces Headache Days in Chronic Migraine: A Randomized, Double-Blind, Placebo-Controlled Study. Cephalalgia 2007, 27, 814–823. [Google Scholar] [CrossRef]
- Diener, H.; Matias-Guiu, J.; Hartung, E.; Pfaffenrath, V.; Ludin, H.; Nappi, G.; de Beukelaar, F. Efficacy and Tolerability in Migraine Prophylaxis of Flunarizine in Reduced Doses: A Comparison with Propranolol 160 Mg Daily. Cephalalgia 2002, 22, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, S.D.; Dodick, D.W.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Fremanezumab for the Preventive Treatment of Chronic Migraine. N. Engl. J. Med. 2017, 377, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Bigal, M.E.; Dodick, D.W.; Rapoport, A.M.; Silberstein, S.D.; Ma, Y.; Yang, R.; Loupe, P.S.; Burstein, R.; Newman, L.C.; Lipton, R.B. Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of high-frequency episodic migraine: A multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol. 2015, 14, 1081–1090. [Google Scholar] [CrossRef]
- Agostoni, E.C.; Barbanti, P.; Calabresi, P.; Colombo, B.; Cortelli, P.; Frediani, F.; Geppetti, P.; Grazzi, L.; Leone, M.; Martelletti, P.; et al. Current and emerging evidence-based treatment options in chronic migraine: A narrative review. J. Headache Pain 2019, 20, 92. [Google Scholar] [CrossRef] [Green Version]
- Akerman, S.; Holland, P.R.; Lasalandra, M.P.; Goadsby, P.J. Endocannabinoids in the Brainstem Modulate Dural Trigeminovascular Nociceptive Traffic via CB1 and “Triptan” Receptors: Implications in Migraine. J. Neurosci. 2013, 33, 14869–14877. [Google Scholar] [CrossRef]
- Sousa-Valente, J.; Varga, A.; Torres-Perez, J.V.; Jenes, A.; Wahba, J.; Mackie, K.; Cravatt, B.; Ueda, N.; Tsuboi, K.; Santha, P.; et al. Inflammation of peripheral tissues and injury to peripheral nerves induce differing effects in the expression of the calcium-sensitive N-arachydonoylethanolamine-synthesizing enzyme and related molecules in rat primary sensory neurons. J. Comp. Neurol. 2017, 525, 1778–1796. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Sominsky, L.; Walder, K.R.; Berk, M.; Marx, W.; Carvalho, A.F.; Bortolasci, C.C.; Maes, M.; Puri, B.K. Inflammation and Nitro-oxidative Stress as Drivers of Endocannabinoid System Aberrations in Mood Disorders and Schizophrenia. Mol. Neurobiol. 2022, 1–19. [Google Scholar] [CrossRef]
- Ahn, K.; McKinney, M.K.; Cravatt, B.F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem. Rev. 2008, 108, 1687–1707. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Luo, L.; Palmer, J.A.; Sutton, S.; Wilson, S.J.; Barbier, A.J.; Breitenbucher, J.G.; Chaplan, S.R.; Webb, M. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br. J. Pharmacol. 2006, 148, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Otrubova, K.; Ezzili, C.; Boger, D.L. The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH). Bioorganic Med. Chem. Lett. 2011, 21, 4674–4685. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, S.G.; Long, J.Z.; O’Neal, S.T.; Abdullah, R.A.; Poklis, J.L.; Boger, D.L.; Cravatt, B.F.; Lichtman, A.H. Blockade of Endocannabinoid-Degrading Enzymes Attenuates Neuropathic Pain. J. Pharmacol. Exp. Ther. 2009, 330, 902–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piomelli, D.; Tarzia, G.; Duranti, A.; Tontini, A.; Mor, M.; Compton, T.R.; Dasse, O.; Monaghan, E.P.; Parrott, J.A.; Putman, D. Pharmacological Profile of the Selective FAAH Inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006, 12, 21–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayamanne, A.; Greenwood, R.; Mitchell, V.A.; Aslan, S.; Piomelli, D.; Vaughan, C.W. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br. J. Pharmacol. 2006, 147, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S. Targeting the Endocannabinoid Metabolic Enzymes to Reduce Inflammatory Pain; ProQuest Dissertations Publishing: Ann Arbor, MI, USA, 2014; p. 174. [Google Scholar]
- Starowicz, K.; Makuch, W.; Korostynski, M.; Malek, N.; Slezak, M.; Zychowska, M.; Petrosino, S.; De Petrocellis, L.; Cristino, L.; Przewlocka, B.; et al. Full Inhibition of Spinal FAAH Leads to TRPV1-Mediated Analgesic Effects in Neuropathic Rats and Possible Lipoxygenase-Mediated Remodeling of Anandamide Metabolism. PLoS ONE 2013, 8, e60040. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Wise, L.E.; Chen, Y.; Gujjar, R.; Mahadevan, A.; Cravatt, B.F.; Lichtman, A.H. The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci. 2013, 92, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Greco, R.; Demartini, C.; Zanaboni, A.; Casini, I.; De Icco, R.; Reggiani, A.; Misto, A.; Piomelli, D.; Tassorelli, C. Characterization of the peripheral FAAH inhibitor, URB937, in animal models of acute and chronic migraine. Neurobiol. Dis. 2021, 147, 105157. [Google Scholar] [CrossRef]
- Greco, R.; Bandiera, T.; Mangione, A.; Demartini, C.; Siani, F.; Nappi, G.; Sandrini, G.; Guijarro, A.; Armirotti, A.; Piomelli, D.; et al. Effects of peripheral FAAH blockade on NTG-induced hyperalgesia—evaluation of URB937 in an animal model of migraine. Cephalalgia 2015, 35, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Korhonen, J.; Kuusisto, A.; Van Bruchem, J.; Patel, J.Z.; Laitinen, T.; Navia-Paldanius, D.; Laitinen, J.T.; Savinainen, J.R.; Parkkari, T.; Nevalainen, T.J. Piperazine and piperidine carboxamides and carbamates as inhibitors of fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL). Bioorganic Med. Chem. 2014, 22, 6694–6705. [Google Scholar] [CrossRef]
- Patel, J.Z.; Parkkari, T.; Laitinen, T.; Kaczor, A.A.; Saario, S.M.; Savinainen, J.R.; Navia-Paldanius, D.; Cipriano, M.; Leppänen, J.; Koshevoy, I.O.; et al. Chiral 1,3,4-Oxadiazol-2-ones as Highly Selective FAAH Inhibitors. J. Med. Chem. 2013, 56, 8484–8496. [Google Scholar] [CrossRef]
- Balapal, S. Basavarajapp Critical Enzymes Involved in Endocannabinoid Metabolism. Protein pept Lett. 2007, 14, 237–246. [Google Scholar] [CrossRef]
- Dinh, T.P.; Freund, T.F.; Piomelli, D. A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem. Phys. Lipids 2002, 121, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Saario, S.M.; Salo, O.M.H.; Nevalainen, T.; Poso, A.; Laitinen, J.T.; Järvinen, T.; Niemi, R. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem. Biol. 2005, 12, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Kano, M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J. Neurosci. 2007, 27, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, A.G.; Suplita, R.L.; Bolton, N.M.; Neely, M.H.; Fegley, D.; Mangieri, R.; Krey, J.F.; Michael Walker, J.; Holmes, P.V.; Crystal, J.D.; et al. An endocannabinoid mechanism for stress-induced analgesia. Nature 2005, 435, 1108–1112. [Google Scholar] [CrossRef]
- Chang, J.W.; Niphakis, M.J.; Lum, K.M.; Cognetta, A.B.; Wang, C.; Matthews, M.L.; Niessen, S.; Buczynski, M.W.; Parsons, L.H.; Cravatt, B.F. Highly Selective Inhibitors of Monoacylglycerol Lipase Bearing a Reactive Group that Is Bioisosteric with Endocannabinoid Substrates. Chem. Biol. 2012, 19, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Berliocchi, L.; Piomelli, D.; Tassorelli, C. Inhibition of monoacylglycerol lipase: Another signalling pathway for potential therapeutic targets in migraine? Cephalalgia 2018, 38, 1138–1147. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Puighermanal, E.; Pastor, A.; De La Torre, R.; Maldonado, R.; Ozaita, A. Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol. Psychiatry 2011, 70, 479–486. [Google Scholar] [CrossRef]
- Spradley, J.M.; Guindon, J.; Hohmann, A.G. Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacol. Res. 2010, 62, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Ignatowska-Jankowska, B.M.; Ghosh, S.; Crowe, M.S.; Kinsey, S.G.; Niphakis, M.J.; Abdullah, R.A.; Tao, Q.; O’Neal, S.T.; Walentiny, D.M.; Wiley, J.L.; et al. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: Antinociceptive activity without cannabimimetic side effects. Br. J. Pharmacol. 2014, 171, 1392–1407. [Google Scholar] [CrossRef] [Green Version]
- Griebel, G.; Pichat, P.; Beeské, S.; Leroy, T.; Redon, N.; Jacquet, A.; Françon, D.; Bert, L.; Even, L.; Lopez-Grancha, M.; et al. Selective blockade of the hydrolysis of the endocannabinoid 2-arachidonoylglycerol impairs learning and memory performance while producing antinociceptive activity in rodents. Sci. Rep. 2015, 5, 7642. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavón, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H.; et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009, 5, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piomelli, D.; Tagne, A.M. Endocannabinoid-Based Therapies. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 483–507. [Google Scholar] [CrossRef] [PubMed]
- Niphakis, M.J.; Cognetta, A.B.; Chang, J.W.; Buczynski, M.W.; Parsons, L.H.; Byrne, F.; Burston, J.J.; Chapman, V.; Cravatt, B.F. Evaluation of NHS Carbamates as a Potent and Selective Class of Endocannabinoid Hydrolase Inhibitors. ACS Chem. Neurosci. 2013, 4, 1322–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatowska-Jankowska, B.; Wilkerson, J.L.; Mustafa, M.; Abdullah, R.; Niphakis, M.; Wiley, J.L.; Cravatt, B.F.; Lichtman, A.H. Selective Monoacylglycerol Lipase Inhibitors: Antinociceptive versus Cannabimimetic Effects in Mice. J. Pharmacol. Exp. Ther. 2015, 353, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.; Wang, Z.; Zhang, Y.; Chen, N. Potential application of endocannabinoid system agents in neuropsychiatric and neurodegenerative diseases—focusing on FAAH/MAGL inhibitors. Acta Pharmacol. Sin. 2020, 41, 1263–1271. [Google Scholar] [CrossRef]
- Aaltonen, N.; Savinainen, J.R.; Ribas, C.R.; Rönkkö, J.; Kuusisto, A.; Korhonen, J.; Navia-Paldanius, D.; Häyrinen, J.; Takabe, P.; Käsnänen, H.; et al. Piperazine and Piperidine Triazole Ureas as Ultrapotent and Highly Selective Inhibitors of Monoacylglycerol Lipase. Chem. Biol. 2013, 20, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Moreira, F.A.; Grieb, M.; Lutz, B. Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: Focus on anxiety and depression. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 133–144. [Google Scholar] [CrossRef]
- Adamson Barnes, N.S.; Mitchell, V.A.; Kazantzis, N.P.; Vaughan, C.W. Actions of the dual FAAH/MAGL inhibitor JZL195 in a murine neuropathic pain model. Br. J. Pharmacol. 2016, 173, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Greco, R.; Demartini, C.; Francavilla, M.; Zanaboni, A.M.; Tassorelli, C. Dual Inhibition of FAAH and MAGL Counteracts Migraine-like Pain and Behavior in an Animal Model of Migraine. Cells 2021, 10, 2543. [Google Scholar] [CrossRef]
- Papa, A.; Pasquini, S.; Contri, C.; Gemma, S.; Campiani, G.; Butini, S.; Varani, K.; Vincenzi, F. Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition. Cells 2022, 11, 471. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J. Recent advances in understanding migraine mechanisms, molecules and therapeutics. Trends Mol. Med. 2007, 13, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, H.; Rahgozar, M.; Speckmann, E.-J.; Gorji, A. Effect of cannabinoid receptor activation on spreading depression. Iran. J. Basic Med. Sci. 2012, 15, 926–936. [Google Scholar] [PubMed]
- Colín-González, A.L.; Aguilera, G.; Santamaría, A. Cannabinoids: Glutamatergic Transmission and Kynurenines. In The Benefits of Natural Products for Neurodegenerative Diseases; Springer: Berlin/Heidelberg, Germany, 2016; pp. 173–198. [Google Scholar]
- Shatillo, A.; Salo, R.A.; Giniatullin, R.; Gröhn, O.H. Involvement of NMDA receptor subtypes in cortical spreading depression in rats assessed by fMRI. Neuropharmacology 2015, 93, 164–170. [Google Scholar] [CrossRef]
- Nagy-Grócz, G.; Zádor, F.; Dvorácskó, S.; Bohár, Z.; Benyhe, S.; Tömböly, C.; Párdutz, Á.; Vécsei, L. Interactions between the Kynurenine and the Endocannabinoid System with Special Emphasis on Migraine. Int. J. Mol. Sci. 2017, 18, 1617. [Google Scholar] [CrossRef] [Green Version]
- Wasilewski, A.; Krajewska, U.; Owczarek, K.; Lewandowska, U.; Fichna, J. Fatty acid amide hydrolase (FAAH) inhibitor PF-3845 reduces viability, migration and invasiveness of human colon adenocarcinoma Colo-205 cell line: An in vitro study. Acta Biochim. Pol. 2017, 64, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Clapper, J.R.; Moreno-Sanz, G.; Russo, R.; Guijarro, A.; Vacondio, F.; Duranti, A.; Tontini, A.; Sanchini, S.; Sciolino, N.R.; Spradley, J.M.; et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat. Neurosci. 2010, 13, 1265–1270. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Pietra, A.; Savinainen, J.; Giniatullin, R. Inhibiting Endocannabinoid Hydrolysis as Emerging Analgesic Strategy Targeting a Spectrum of Ion Channels Implicated in Migraine Pain. Int. J. Mol. Sci. 2022, 23, 4407. https://doi.org/10.3390/ijms23084407
Della Pietra A, Savinainen J, Giniatullin R. Inhibiting Endocannabinoid Hydrolysis as Emerging Analgesic Strategy Targeting a Spectrum of Ion Channels Implicated in Migraine Pain. International Journal of Molecular Sciences. 2022; 23(8):4407. https://doi.org/10.3390/ijms23084407
Chicago/Turabian StyleDella Pietra, Adriana, Juha Savinainen, and Rashid Giniatullin. 2022. "Inhibiting Endocannabinoid Hydrolysis as Emerging Analgesic Strategy Targeting a Spectrum of Ion Channels Implicated in Migraine Pain" International Journal of Molecular Sciences 23, no. 8: 4407. https://doi.org/10.3390/ijms23084407