
Contribution of Whole-Genome Sequencing and Transcript Analysis to Decipher Retinal Diseases Associated with MFSD8 Variants

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Patients Description
2.2. Genotyping and Transcript Analysis
3. Discussion
3.1. Common or Specific Clinical Features
3.2. Novel Splice Defects
3.3. Genotype–Phenotype Correlations
4. Materials and Methods
4.1. Cohort of Patients
4.2. Clinical Examination
4.3. Genetic Assessment
4.4. Copy Number Variants (CNVs)
4.5. Variant Pathogenicity Assessment
4.6. Lymphoblastoid Cell Lines’ (LCLs) Production
4.7. Transcript Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Georgiou, M.; Kane, T.; Tanna, P.; Bouzia, Z.; Singh, N.; Kalitzeos, A.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Prospective Cohort Study of Childhood-Onset Stargardt Disease: Fundus Autofluorescence Imaging, Progression, Comparison with Adult-Onset Disease, and Disease Symmetry. Am. J. Ophthalmol. 2020, 211, 159–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosing, S.; Thiadens, A.A.H.J.; Hoyng, C.B.; Klaver, C.C.W.; den Hollander, A.I.; Cremers, F.P.M. Causes and consequences of inherited cone disorders. Prog. Retin. Eye Res. 2014, 42, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboshiha, J.; Dubis, A.M.; Carroll, J.; Hardcastle, A.J.; Michaelides, M. The cone dysfunction syndromes. Br. J. Ophthalmol. 2016, 100, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ávila-Fernández, A.; Cantalapiedra, D.; Aller, E.; Vallespín, E.; Aguirre-Lambán, J.; Blanco-Kelly, F.; Corton, M.; Riveiro-Álvarez, R.; Allikmets, R.; Trujillo-Tiebas, M.J.; et al. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol. Vis. 2010, 16, 2550–2558. [Google Scholar] [PubMed]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands With Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef] [PubMed]
- Goetz, K.E.; Reeves, M.J.; Gagadam, S.; Blain, D.; Bender, C.; Lwin, C.; Naik, A.; Tumminia, S.J.; Hufnagel, R.B. Genetic testing for inherited eye conditions in over 6000 individuals through the eyeGENE network. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 828–837. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Koenekoop, R.K.; Senechal, A.; De Baere, E.B.W.; de Ravel, T.; Banfi, S.; Kohl, S.; Ayuso, C.; Sharon, D.; Hoyng, C.B.; et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch. Ophthalmol. Chic. Ill 1960 2012, 130, 1425–1432. [Google Scholar] [CrossRef]
- Chen, F.K.; Zhang, X.; Eintracht, J.; Zhang, D.; Arunachalam, S.; Thompson, J.A.; Chelva, E.; Mallon, D.; Chen, S.-C.; McLaren, T.; et al. Clinical and molecular characterization of non-syndromic retinal dystrophy due to c.175G>A mutation in ceroid lipofuscinosis neuronal 3 (CLN3). Doc. Ophthalmol. Adv. Ophthalmol. 2019, 138, 55–70. [Google Scholar] [CrossRef]
- Smirnov, V.M.; Nassisi, M.; Solis Hernandez, C.; Méjécase, C.; El Shamieh, S.; Condroyer, C.; Antonio, A.; Meunier, I.; Andrieu, C.; Defoort-Dhellemmes, S.; et al. Retinal Phenotype of Patients with Isolated Retinal Degeneration Due to CLN3 Pathogenic Variants in a French Retinitis Pigmentosa Cohort. JAMA Ophthalmol. 2021, 139, 278–291. [Google Scholar] [CrossRef]
- Magliyah, M.S.; Geuer, S.; Alsalamah, A.K.; Lenzner, S.; Drasdo, M.; Schatz, P. Association of the Recurrent Rare Variant c.415T>C p.Phe139Leu in CLN5 with a Recessively Inherited Macular Dystrophy. JAMA Ophthalmol. 2021, 139, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; van den Born, L.I.; Sangermano, R.; Banfi, S.; Koenekoop, R.K.; Zonneveld-Vrieling, M.N.; Klaver, C.C.W.; van Lith-Verhoeven, J.J.C.; Cremers, F.P.M.; den Hollander, A.I.; et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology 2015, 122, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; El-Asrag, M.E.; Ku, C.A.; Holder, G.E.; McKibbin, M.; Arno, G.; Poulter, J.A.; Carss, K.; Bommireddy, T.; Bagheri, S.; et al. Specific Alleles of CLN7/MFSD8, a Protein That Localizes to Photoreceptor Synaptic Terminals, Cause a Spectrum of Nonsyndromic Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2906–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [Green Version]
- Zare-Abdollahi, D.; Bushehri, A.; Alavi, A.; Dehghani, A.; Mousavi-Mirkala, M.; Effati, J.; Miratashi, S.A.M.; Dehani, M.; Jamali, P.; Khorram Khorshid, H.R. MFSD8 gene mutations; evidence for phenotypic heterogeneity. Ophthalmic. Genet. 2019, 40, 141–145. [Google Scholar] [CrossRef]
- Bauwens, M.; Storch, S.; Weisschuh, N.; Ceuterick-de Groote, C.; De Rycke, R.; Guillemyn, B.; De Jaegere, S.; Coppieters, F.; Van Coster, R.; Leroy, B.P.; et al. Functional characterization of novel MFSD8 pathogenic variants anticipates neurological involvement in juvenile isolated maculopathy. Clin. Genet. 2020, 97, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Q.; Cao, Y.; Xu, H.; Yang, Z.; Tang, L.; Xiang, J.; Li, J.; Deng, H.; Yuan, L. Novel MFSD8 Variants in a Chinese Family with Nonsyndromic Macular Dystrophy. J. Ophthalmol. 2021, 2021, 6684045. [Google Scholar] [CrossRef]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 2005, 6, 107–126. [Google Scholar] [CrossRef]
- Jalanko, A.; Braulke, T. Neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta 2009, 1793, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [Green Version]
- Mandel, H.; Cohen Katsanelson, K.; Khayat, M.; Chervinsky, I.; Vladovski, E.; Iancu, T.C.; Indelman, M.; Horovitz, Y.; Sprecher, E.; Shalev, S.A.; et al. Clinico-pathological manifestations of variant late infantile neuronal ceroid lipofuscinosis (vLINCL) caused by a novel mutation in MFSD8 gene. Eur. J. Med. Genet. 2014, 57, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Steenhuis, P.; Herder, S.; Gelis, S.; Braulke, T.; Storch, S. Lysosomal targeting of the CLN7 membrane glycoprotein and transport via the plasma membrane require a dileucine motif. Traffic 2010, 11, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, A.; Kousi, M.; Sagné, C.; Bellenchi, G.C.; Morel, L.; Darmon, M.; Hulková, H.; Ruivo, R.; Debacker, C.; El Mestikawy, S.; et al. Expression and lysosomal targeting of CLN7, a major facilitator superfamily transporter associated with variant late-infantile neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2010, 19, 4497–4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenhuis, P.; Froemming, J.; Reinheckel, T.; Storch, S. Proteolytic cleavage of the disease-related lysosomal membrane glycoprotein CLN7. Biochim. Biophys. Acta 2012, 1822, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Kleist, L.; Ariunbat, K.; Braren, I.; Stauber, T.; Storch, S.; Danyukova, T. A newly generated neuronal cell model of CLN7 disease reveals aberrant lysosome motility and impaired cell survival. Mol. Genet. Metab. 2019, 126, 196–205. [Google Scholar] [CrossRef]
- Huber, R.J.; Mathavarajah, S.; Yap, S.Q. Mfsd8 localizes to endocytic compartments and influences the secretion of Cln5 and cathepsin D in Dictyostelium. Cell. Signal. 2020, 70, 109572. [Google Scholar] [CrossRef]
- Reith, M.; Zeltner, L.; Schäferhoff, K.; Witt, D.; Zuleger, T.; Haack, T.B.; Bornemann, A.; Alber, M.; Ruf, S.; Schoels, L.; et al. A Novel, Apparently Silent Variant in MFSD8 Causes Neuronal Ceroid Lipofuscinosis with Marked Intrafamilial Variability. Int. J. Mol. Sci. 2022, 23, 2271. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [Green Version]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Lambertus, S.; van Huet, R.A.C.; Bax, N.M.; Hoefsloot, L.H.; Cremers, F.P.M.; Boon, C.J.F.; Klevering, B.J.; Hoyng, C.B. Early-onset stargardt disease: Phenotypic and genotypic characteristics. Ophthalmology 2015, 122, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Zernant, J.; Chana, R.K.; Wright, G.A.; Tsunoda, K.; Ozawa, Y.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Allikmets, R.; et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 2015, 122, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am. J. Ophthalmol. 2013, 156, 487–501.e1. [Google Scholar] [CrossRef] [PubMed]
- Westeneng-van Haaften, S.C.; Boon, C.J.F.; Cremers, F.P.M.; Hoefsloot, L.H.; den Hollander, A.I.; Hoyng, C.B. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 2012, 119, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Nõupuu, K.; Lee, W.; Zernant, J.; Tsang, S.H.; Allikmets, R. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7217–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, N.M.; Lambertus, S.; Cremers, F.P.M.; Klevering, B.J.; Hoyng, C.B. The absence of fundus abnormalities in Stargardt disease. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Lois, N.; Holder, G.E.; Bunce, C.; Fitzke, F.W.; Bird, A.C. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch. Ophthalmol. Chic. Ill 1960 2001, 119, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, K.; Lois, N.; Davidson, A.E.; Mackay, D.S.; Hogg, C.R.; Stone, E.M.; Tsunoda, K.; Tsubota, K.; Bunce, C.; Robson, A.G.; et al. A longitudinal study of stargardt disease: Clinical and electrophysiologic assessment, progression, and genotype correlations. Am. J. Ophthalmol. 2013, 155, 1075–1088.e13. [Google Scholar] [CrossRef]
- Boon, C.J.F.; Jeroen Klevering, B.; Keunen, J.E.E.; Hoyng, C.B.; Theelen, T. Fundus autofluorescence imaging of retinal dystrophies. Vis. Res. 2008, 48, 2569–2577. [Google Scholar] [CrossRef] [Green Version]
- Lois, N.; Halfyard, A.S.; Bird, A.C.; Holder, G.E.; Fitzke, F.W. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am. J. Ophthalmol. 2004, 138, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Sumaroka, A.; Schwartz, S.B.; Roman, M.I.; Milam, A.H.; Bennett, J.; Stone, E.M.; Jacobson, S.G. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4739–4746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Weleber, R.G.; Michaelides, M.; Trzupek, K.M.; Stover, N.B.; Stone, E.M. The phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Preising, M.N.; Abura, M.; Jäger, M.; Wassill, K.-H.; Lorenz, B. Ocular morphology and function in juvenile neuronal ceroid lipofuscinosis (CLN3) in the first decade of life. Ophthalmic. Genet. 2017, 38, 252–259. [Google Scholar] [CrossRef]
- Wright, G.A.; Georgiou, M.; Robson, A.G.; Ali, N.; Kalhoro, A.; Holthaus, S.K.; Pontikos, N.; Oluonye, N.; de Carvalho, E.R.; Neveu, M.M.; et al. Juvenile Batten Disease (CLN3): Detailed Ocular Phenotype, Novel Observations, Delayed Diagnosis, Masquerades, and Prospects for Therapy. Ophthalmol. Retin. 2019, 4, 433–445. [Google Scholar] [CrossRef]
- Orlin, A.; Sondhi, D.; Witmer, M.T.; Wessel, M.M.; Mezey, J.G.; Kaminsky, S.M.; Hackett, N.R.; Yohay, K.; Kosofsky, B.; Souweidane, M.M.; et al. Spectrum of ocular manifestations in CLN2-associated batten (Jansky-Bielschowsky) disease correlate with advancing age and deteriorating neurological function. PLoS ONE 2013, 8, e73128. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Tomkiewicz, T.Z.; Suárez-Herrera, N.; Cremers, F.P.M.; Collin, R.W.J.; Garanto, A. Antisense Oligonucleotide-Based Rescue of Aberrant Splicing Defects Caused by 15 Pathogenic Variants in ABCA4. Int. J. Mol. Sci. 2021, 22, 4621. [Google Scholar] [CrossRef]
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.-Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.-K.; et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 2007, 81, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Tubeuf, H.; Charbonnier, C.; Soukarieh, O.; Blavier, A.; Lefebvre, A.; Dauchel, H.; Frebourg, T.; Gaildrat, P.; Martins, A. Large-scale comparative evaluation of user-friendly tools for predicting variant-induced alterations of splicing regulatory elements. Hum. Mutat. 2020, 41, 1811–1829. [Google Scholar] [CrossRef]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A.; et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 2020, 11, 3328. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wu, N.; Zaneveld, S.; Liu, H.; Fu, S.; Wang, K.; Bertrand, R.; Wang, J.; Li, Y.; Chen, R. Transcript isoforms of Reep6 have distinct functions in the retina. Hum. Mol. Genet. 2021, 30, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Mathur, P.D.; Zou, J.; Zheng, T.; Almishaal, A.; Wang, Y.; Chen, Q.; Wang, L.; Vashist, D.; Brown, S.; Park, A.; et al. Distinct expression and function of whirlin isoforms in the inner ear and retina: An insight into pathogenesis of USH2D and DFNB31. Hum. Mol. Genet. 2015, 24, 6213–6228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, C.; Terracciano, A.; Simonati, A.; Discepoli, G.; Cannelli, N.; Claps, D.; Crow, Y.J.; Bianchi, M.; Kitzmuller, C.; Longo, D.; et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 2009, 30, E530–E540. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Siintola, E.; Dvorakova, L.; Vlaskova, H.; Turnbull, J.; Topcu, M.; Yuksel, D.; Gokben, S.; Minassian, B.A.; Elleder, M.; et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain J. Neurol. 2009, 132, 810–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheldof, A.; Seneca, S.; Stouffs, K.; Lissens, W.; Jansen, A.; Laeremans, H.; Verloo, P.; Schoonjans, A.-S.; Meuwissen, M.; Barca, D.; et al. Clinical implementation of gene panel testing for lysosomal storage diseases. Mol. Genet. Genomic. Med. 2019, 7, e00527. [Google Scholar] [CrossRef]
- Craiu, D.; Dragostin, O.; Dica, A.; Hoffman-Zacharska, D.; Gos, M.; Bastian, A.E.; Gherghiceanu, M.; Rolfs, A.; Nahavandi, N.; Craiu, M.; et al. Rett-like onset in late-infantile neuronal ceroid lipofuscinosis (CLN7) caused by compound heterozygous mutation in the MFSD8 gene and review of the literature data on clinical onset signs. Eur. J. Paediatr. Neurol. 2015, 19, 78–86. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Al-Hassnan, Z.N.; Aldosari, M.; Alkuraya, F.S. Neuronal Ceroid Lipofuscinosis Caused by MFSD8 Mutations: A Common Theme Emerging. Neurogenetics 2009, 10, 307–311. [Google Scholar] [CrossRef]
- Patiño, L.C.; Battu, R.; Ortega-Recalde, O.; Nallathambi, J.; Anandula, V.R.; Renukaradhya, U.; Laissue, P. Exome Sequencing Is an Efficient Tool for Variant Late-Infantile Neuronal Ceroid Lipofuscinosis Molecular Diagnosis. PLoS ONE 2014, 9, e109576. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.; Grunewald, O.; Muller, J.; Zeitz, C.; Obermaier, C.D.; Devos, A.; Pelletier, V.; Bocquet, B.; Andrieu, C.; Bacquet, J.-L.; et al. Novel TTLL5 Variants Associated with Cone-Rod Dystrophy and Early-Onset Severe Retinal Dystrophy. Int. J. Mol. Sci. 2021, 22, 6410. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID, Sex | Age at Initial Examination | Symptoms | BCVA at First Examination | Refraction | Fundus | Colour Vision | Visual Field | ERG | SW-FAF | SD-OCT | Retinal Phenotype |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Syndromic IRD in Late-Infantile Ceroid Lipofuscinosis | |||||||||||
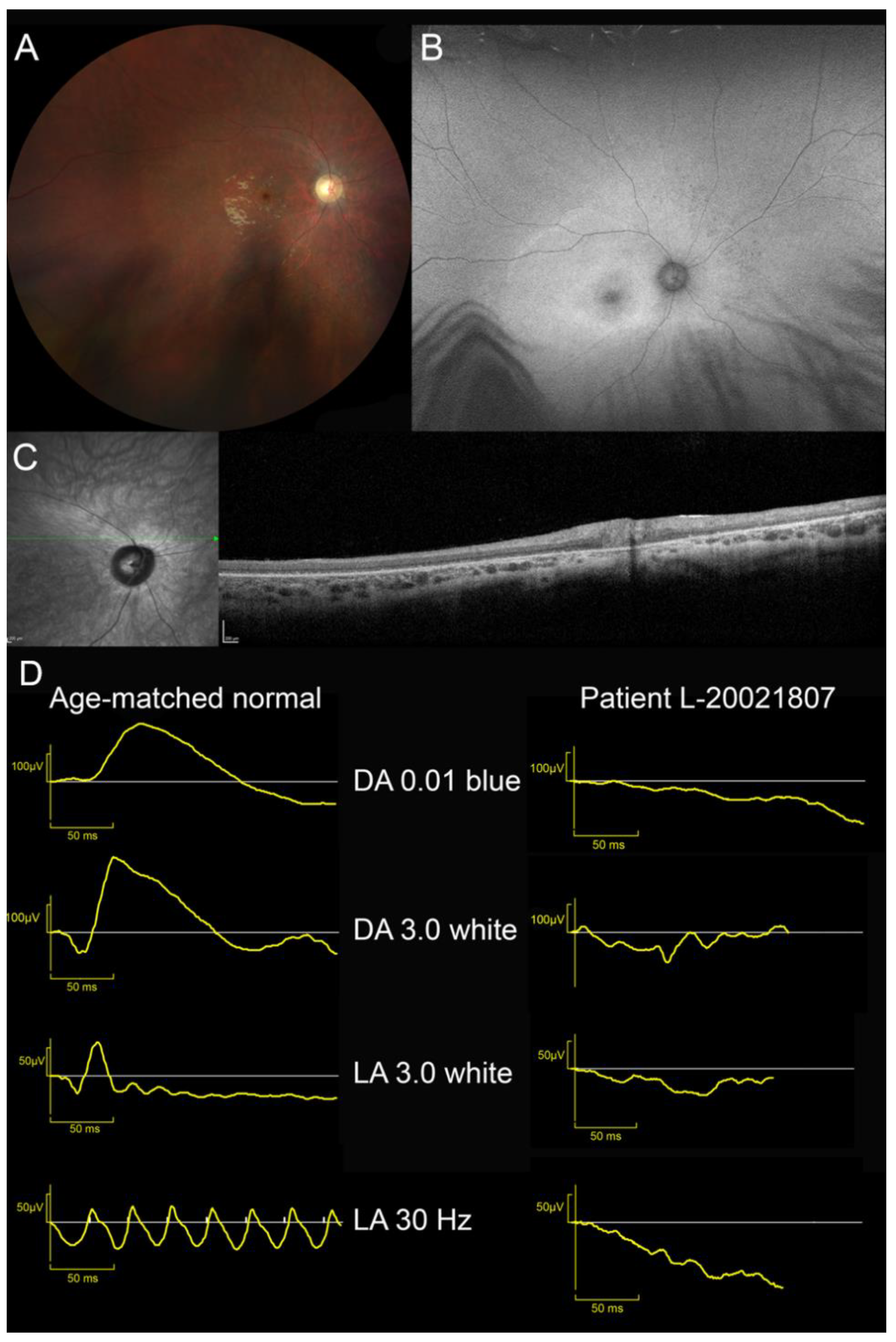
| L-20021807, M | 10 | VA loss and photophobia | 20/400 OU | Cyclopleged +1.50 OU | Waxy pallor of ONH Severe vascular attenuation Yellowish macula Cellophane light reflex | NA | 20° central scotoma at III3e target V4e normal | Unrecordable | Large area of increased autofluorescence with indistinct border in the posterior pole and the second more narrow annulus of increased autofluorescence around the fovea | Overall retinal thinning; Indistinct lamination and disappearance of outer layers (EZ and RPE) | EOSRD |
| Isolated IRD | |||||||||||
| Teenage-onset forms | |||||||||||
| L-08031428, M | 12 | VA loss, poor colour discrimination and photophobia | 20/125 OU | Cyclopleged +1.25 (−0.50) 155° RE +1.75 (−0.75) 30° LE | Temporal pallor of ONH Arteriolar narrowing Punched-out round foveal lesion with hyperpigmented edges | Severe without-axis dyschromatopsia on Lantony D-15 | Small relative central scotoma on static VF; Kinetic VF normal to all target sizes | Cone–rod dysfunction Reduced b/a ratio in DA 3.0 ERG | Round foveal hypoAF lesion | Foveal outer layer cavitation | CORD |
| IM-190703, M | 12 | VA loss, reading difficulties | 20/32 OU | (−0.75) 90° RE −0.75 (−0.5) 90° LE | Temporal pallor of ONH Punched-out round foveal lesion with hyperpigmented edges | NA | Central scotoma | Scotopic normal Photopic reduced to 1/3 normal limits | Round hypoAF lesion | Loss of foveal outer reflective layers (ONL, EZ and RPE) | COD |
| VV-1595021, F | 14 | VA loss, reading difficulties, photophobia | 20/40 OU | +0.75 OU | Punched out round foveal lesion with hyperpigmented edges | Ishihara normal | 10° relative central scotoma at I1e target on static VF | Scotopic/photopic within normal limits | Round hypoAF lesion | Foveal outer layer cavitation | MD |
| Adult-onset forms | |||||||||||
| HD-OPH1206, M | 30 | VA loss, photophobia (especially outdoors), contrast issues | 20/63 RE 20/80 LE | −0.25 (−0.50) 170° RE −0.50 (−0.25) 80° LE | Punched-out round macular lesion with hyperpigmented edges | Severe without-axis dyschromatopsia on Lantony D-15 | 5° relative central scotoma at I1e target on static VF | Scotopic within normal limits Photopic reduced | Round hypoAF lesion with watershade edges | Loss of macular outer reflective layers (ONL, EZ and RPE) | COD |
| HD-OPH4200, F | 37 | VA loss, reading difficulties, asthenopia, reduction in contrast sensitivity | 20/50 RE 20/32 LE | −1.75 (−0.25) 177° RE −2.00 (−0.25) 159° LE | Temporal pallor of ONH Loss of foveal light reflex | Red–green dyschromatopsia with the Farnsworth 100 Hue | 5° relative central scotoma at I1e target on static VF | Scotopic within normal limits Photopic reduced | Round hypoAF lesion, surrounded by hyperAF edges | Loss of macular outer reflective layers (ONL, EZ and RPE) | COD |
| VV-51717, M | 55 | VA loss, reading difficulties, photophobia | 20/100 OU | −0.50 (−0.50) 0° RE −0.75 (−0.50) 0° LE | Temporal pallor of ONH, Punched-out round macular lesion with hyperpigmented edges | Ishihara normal | NA | Scotopic/photopic within normal limits | Round hypoAF lesion, surrounded by hyperAF edges | Loss of macular outer reflective layers (ONL, EZ and RPE) | MD |
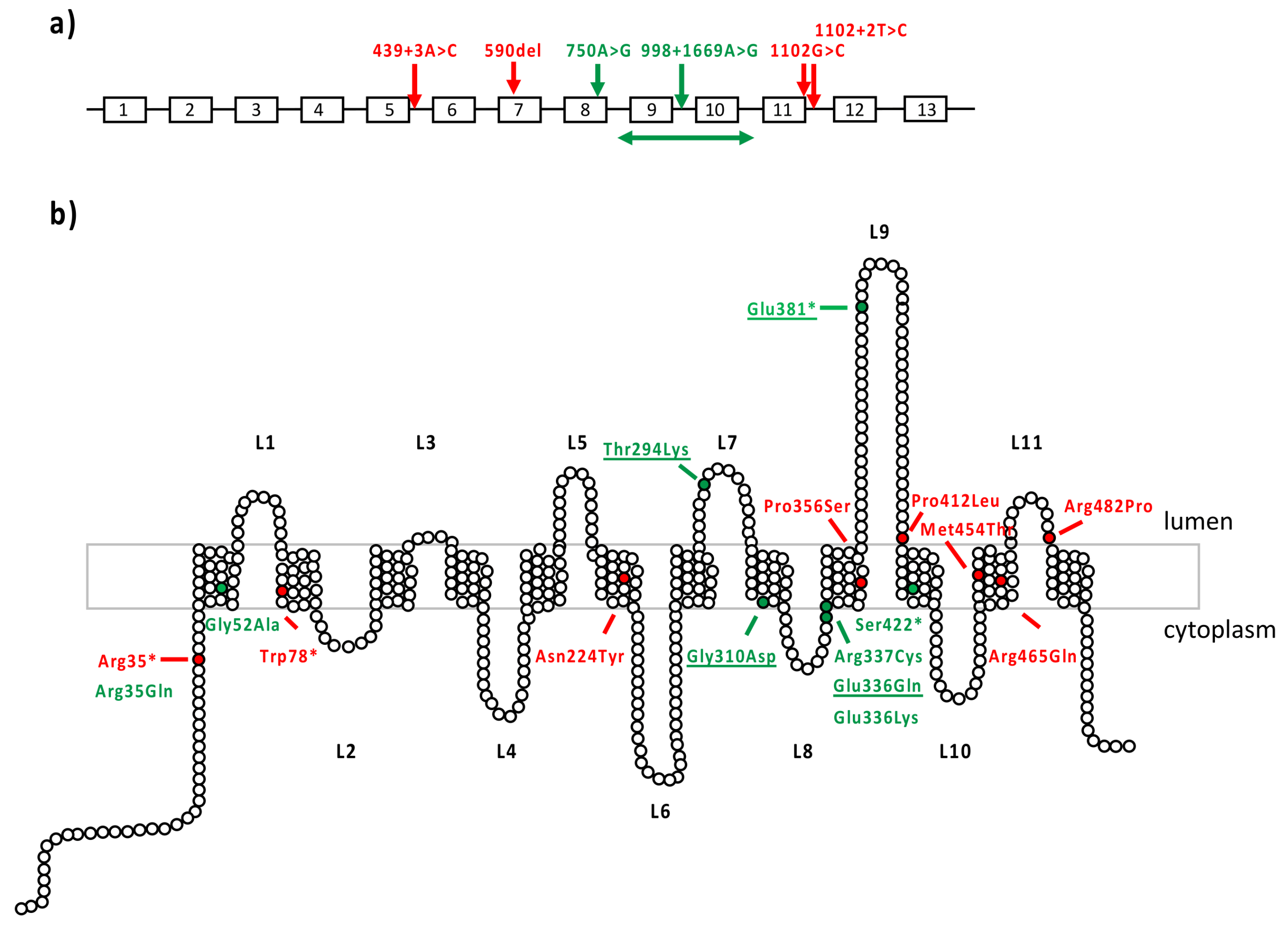
| Patient ID | Phenotype | Genomics Position (Hg19) | DNA Variant | Protein Variant | Variant Type | gnomAD AF | In Silico Prediction | Grantham Distance | ACMG Classification | Reference/ Phenotype |
|---|---|---|---|---|---|---|---|---|---|---|
| Syndromic IRD | ||||||||||
| L-20021807 | LINCL/ EOSRD | Chr4:128859942 | c.750A>G | p.[Arg233Serfs*5,=] | Synonymous/Splicing | 0 | Exon 8 skipping SpliceAI: MFSD8|0.00|0.15|0.00|0.45|-4|49|0|-4 | - | Likely Pathogenic (PS3,PM3,PP3,PP5) | Reith 2022 LINCL |
| Chr4:128849857–128856974 | c.(755-2726_998+1981)delinsGTA | p.(Ser253Leufs*79) | Copy Number Variation | 0 | Multiexons deletion (ex9-10) | - | Pathogenic (PVS1,PS3,PM2) | This study | ||
| Isolated IRD | ||||||||||
| L-08031428 | CORD | Chr4:128859942 | c.750A>G | p.[Arg233Serfs*5,=] | Synonymous/Splicing | 0 | Exon 8 skipping SpliceAI: MFSD8|0.00|0.15|0.00|0.45|-4|49|0|-4 | - | Likely Pathogenic (PS3,PM3,PP3,PP5) | Reith 2022 LINCL |
| Chr4:128843111 | c.1006G>A | p.(Glu336Lys) | Missense | 0.00000882 | CADD: 29.9 | 56 | Likely Pathogenic (PM2,PM3,PM5,PP3) | This study | ||
| IM-190703 | COD | Chr4:128878706 Chr4:128871002 | c.104G>A* c.155G>C* | p.(Arg35Gln) p.(Gly52Ala) | Missense Missense | 0.00000797 0 | CADD: 28.0 CADD: 24,9 | 43 60 | Unknown significance (PM2,PM3,PP3) Likely Pathogenic ((PM2,PM3,PM5,PP3) | This study |
| This study | ||||||||||
| Chr4:128842764 | c.1265C>A | p.(Ser422*) | Nonsense | 0 | CADD: 37.0 | - | Pathogenic (PVS1,PM2) | This study | ||
| VV-1595021 | MD | Chr4:128843108 | c.1009C>T | p.(Arg337Cys) | Missense | 0.00001195 | CADD: 28.6 | 180 | Likely Pathogenic (PM2-S,PM3,PP3) | This study |
| Chr4:128842888 | c.1141G>T | p.(Glu381*) | Nonsense | 0.000007075 | CADD: 41.0 | - | Pathogenic (PVS1,PM2,PP5-S) | Roosing 2015 MD | ||
| HD-OPH1206 | COD | Chr4:128851907 | c.929G>A | p.(Gly310Asp) | Missense | 0 | CADD: 23.9 | 94 | Pathogenic (PP5-S,PM2,PP3) | Siintola 2007 LINCL |
| Chr4:128850169 | c.998+1669A>G | p.[=,Lys333Asnfs*18] | Deep Intronic Variation | 0 | 140 bp pseudoexon insertion, SpliceAI: MFSD8|0.30|0.00|0.68|0.13|140|-3|1|-3 | - | Likely Pathogenic (PS3,PM3,PM2,PP3) | This study | ||
| HD-OPH4200 | COD | Chr4:128851955 | c.881C>A | p.(Thr294Lys) | Missense | 0.00000884 | CADD: 24.4 | 78 | Likely Pathogenic (PP5-VS,PM2-S,PP3) | Aiello 2009 LINCL |
| Chr4:128843111 | c.1006G>C | p.(Glu336Gln) | Missense | 0.00301 | CADD: 24.2 | 29 | Likely Pathogenic (PS4,PP5-M,PP3,BS1) | Roosing 2015 MD | ||
| VV-51717 | MD | Chr4:128843111 | c.1006G>C | p.(Glu336Gln) | Missense | 0.002494 | CADD: 24.2 | 29 | Likely Pathogenic (PS4,PP5-M,PP3,BS1) | Roosing 2015 MD |
| Chr4:128851955 | c.881C>A | p.(Thr294Lys) | Missense | 0.000008018 | CADD: 24.4 | 78 | Likely Pathogenic (PP5-VS,PM2-S,PP3) | Aiello 2009 LINCL | ||
| Isolated Retinal Degeneration | Late-Infantile Neuronal Ceroid Lipofuscinosis | ||||||
|---|---|---|---|---|---|---|---|
| Allele 1 | Predicted Effect | Allele 2 | Predicted Effect | Reference | Allele 2 | Predicted Effect | Reference |
| c.750A>G p.[Arg233Serfs*5,=] | severe (exon skipping) | c.1006G>A p.(Glu336Lys) | mild (conservative substitution) | This study | c.(755-2726_998+1981)delinsGTA p.(Ser253Leufs*79) | severe (truncating) | This study |
| c.750A>G p.[Arg233Serfs*5,=] | severe (exon skipping) | [27] | |||||
| c.881C>A p.(Thr294Lys) | severe (enhanced proteolytic cleavage) [24] | c.1006G>C p.(Glu336Gln) | mild (conservative substitution) | This study (2 cases) | c.881C>A p.(Thr294Lys) | severe (enhanced proteolytic cleavage) [24] | [55] [56] (18 cases) [57] |
| c.754+2T>A p.(?) | severe (splice variant) | [58] | |||||
| c.929G>A p.(Gly310Glu) | severe (moderately conservative substitution) | c.998+1669A>G p.[=,Lys333Asnfs*18] | moderate (wt transcript 50%) | This study | c.863+3_4insT p.(?) | severe (splice variant) | [55] |
| c.929G>A p.(Gly310Glu) | severe (moderately conservative substitution) | [50] | |||||
| c.1006G>C p.(Glu336Gln) | mild (conservative substitution) | c.1141G>T p.(Glu381*) | severe (truncating) | [2] | |||
| c.1102G>C p.(Lys333Lysfs*3) | severe (truncating) | [2] | |||||
| c.103C>T p.(Arg35*) | severe (truncating) | [13] | |||||
| c.1394G>A p.(Arg465Gln) | supposed severe (although conservative substitution) | [13] | |||||
| c.233G>A p.(Trp78*) | severe (truncating) | [13] | |||||
| c.881C>A p.(Thr294Lys) | severe (enhanced proteolytic cleavage) [24] | This study (2 cases) | |||||
| c.1066C>T p.(Pro356Ser) | moderate (moderately conservative substitution) | c.1102+2T>C p.(?) | severe (splice variant) | [17] | |||
| c.1141G>T p.(Glu381*) | severe (truncating) | c.1006G>C p.(Glu336Gln) | mild (conservative substitution) | [2] | |||
| c.1009C>T p.(Arg337Cys) | moderate (radical substitution) | This study | |||||
| c.1235C>T p.(Pro412Leu) | severe (increased proteolytic cleavage) [24] | c.1361T>C p.(Met454Thr) | moderate (moderately conservative substitution) | [13] (6 families) [15] | c.1235C>T p.(Pro412Leu) | severe (increased proteolytic cleavage) [24] | [59] |
| c.1265C>A p.(Ser422*) | severe (truncating) | c.[104G>A;155G>C] p. [(Arg35Gln;Gly52Ala] | mild (conservative substitution) | This study | |||
| c.1361T>C p.(Met454Thr) | moderate (moderately conservative substitution) | c.1361T>C p.(Met454Thr) | moderate (moderately conservative substitution) | [13] (6 families) [15] | in cis with c.1219T>C p.(Trp407Arg) at the homozygous state | severe (moderately radical) | [60] |
| c.1235C>T p.(Pro412Leu) | severe (increased proteolytic cleavage) [24] | [15] | |||||
| c.1394G>A p.(Arg465Gln) | supposed severe (although conservative substitution) | c.1006G>C p.(Glu336Gln) | mild (conservative substitution) | [13] | c.1394G>A p.(Arg465Gln) | supposed severe (although conservative substitution) | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poncet, A.F.; Grunewald, O.; Vaclavik, V.; Meunier, I.; Drumare, I.; Pelletier, V.; Bocquet, B.; Todorova, M.G.; Le Moing, A.-G.; Devos, A.; et al. Contribution of Whole-Genome Sequencing and Transcript Analysis to Decipher Retinal Diseases Associated with MFSD8 Variants. Int. J. Mol. Sci. 2022, 23, 4294. https://doi.org/10.3390/ijms23084294
Poncet AF, Grunewald O, Vaclavik V, Meunier I, Drumare I, Pelletier V, Bocquet B, Todorova MG, Le Moing A-G, Devos A, et al. Contribution of Whole-Genome Sequencing and Transcript Analysis to Decipher Retinal Diseases Associated with MFSD8 Variants. International Journal of Molecular Sciences. 2022; 23(8):4294. https://doi.org/10.3390/ijms23084294
Chicago/Turabian StylePoncet, Anaïs F., Olivier Grunewald, Veronika Vaclavik, Isabelle Meunier, Isabelle Drumare, Valérie Pelletier, Béatrice Bocquet, Margarita G. Todorova, Anne-Gaëlle Le Moing, Aurore Devos, and et al. 2022. "Contribution of Whole-Genome Sequencing and Transcript Analysis to Decipher Retinal Diseases Associated with MFSD8 Variants" International Journal of Molecular Sciences 23, no. 8: 4294. https://doi.org/10.3390/ijms23084294