
Autophagy Signaling by Neural-Induced Human Adipose Tissue-Derived Stem Cell-Conditioned Medium during Rotenone-Induced Toxicity in SH-SY5Y Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effects of NI-hADSC-CM on ROT-Induced ROS Formation in SH-SY5Y Cells
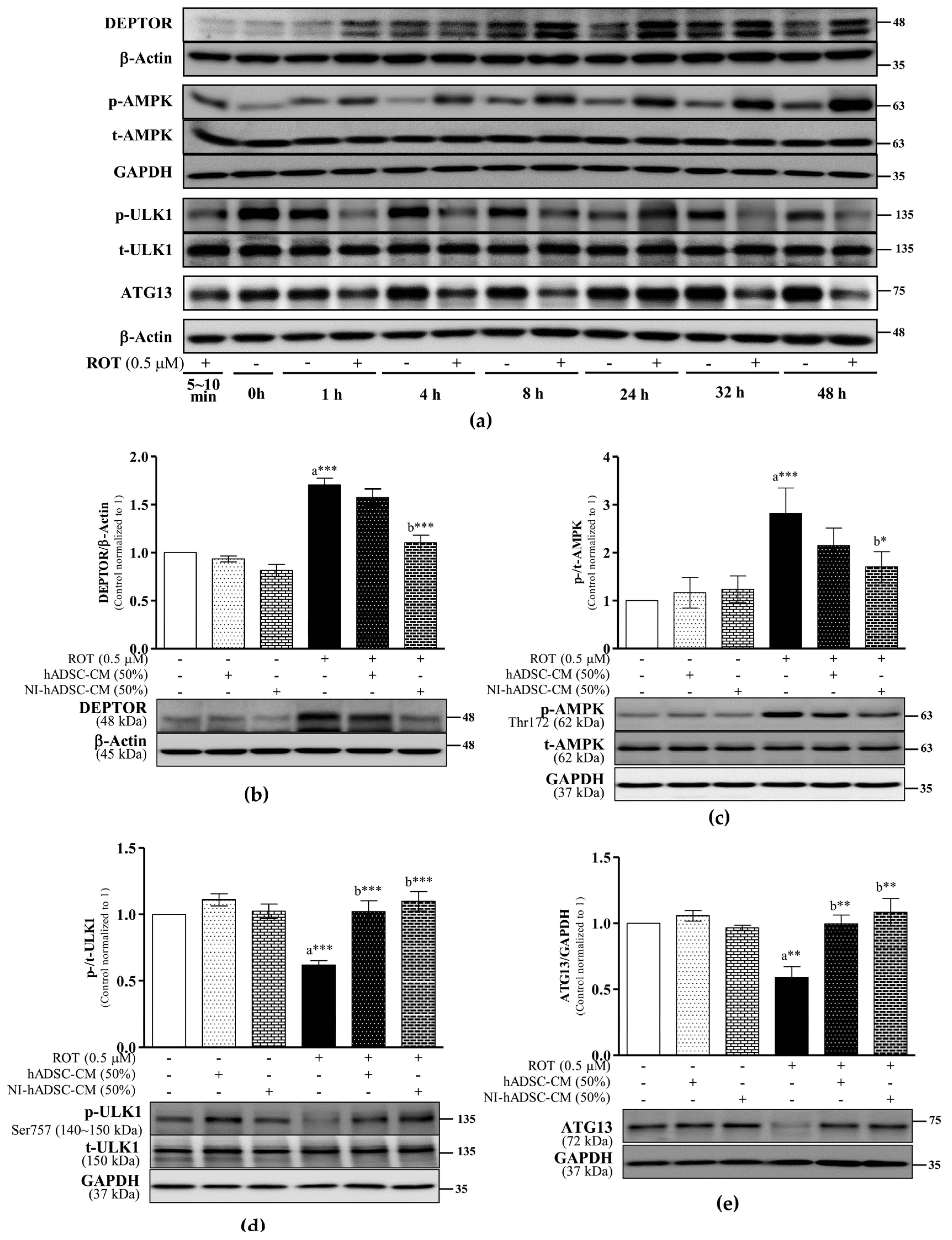
2.2. Effects of NI-hADSC-CM on mTORC1, mTORC2, and DEPTOR Protein Expression in ROT-Exposed SH-SY5Y Cells
2.3. Effects of NI-hADSC-CM on AMPK, ULK1, and ATG13 Protein Expression in ROT-Exposed SH-SY5Y Cells
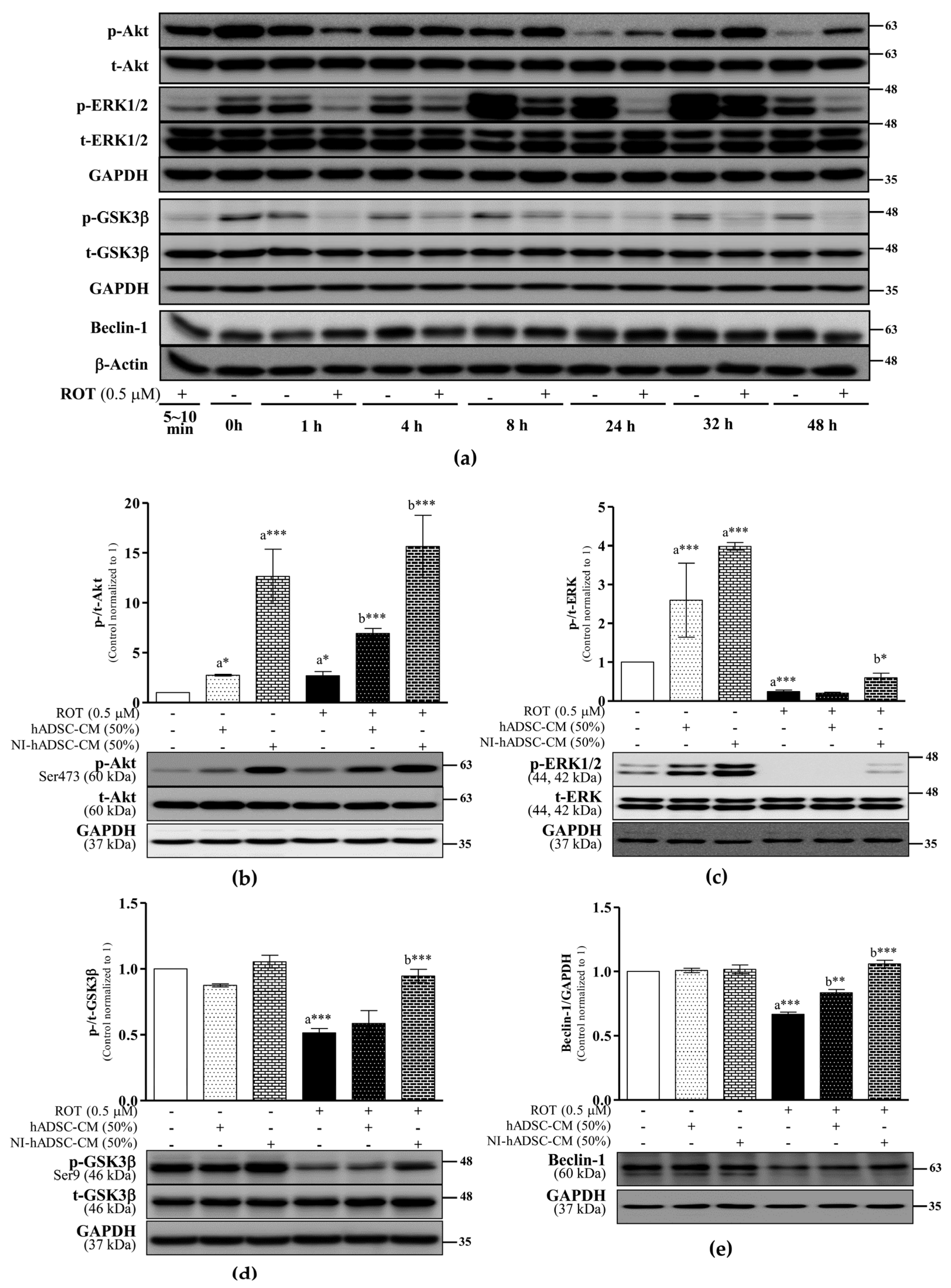
2.4. Effects of NI-hADSC-CM on Akt, ERK, GSK3β, and Beclin-1 Protein Expression in ROT-Exposed SH-SY5Y Cells
2.5. Effects of NI-hADSC-CM on LC3B and p62 Protein Expression in ROT-Exposed SH-SY5Y Cells
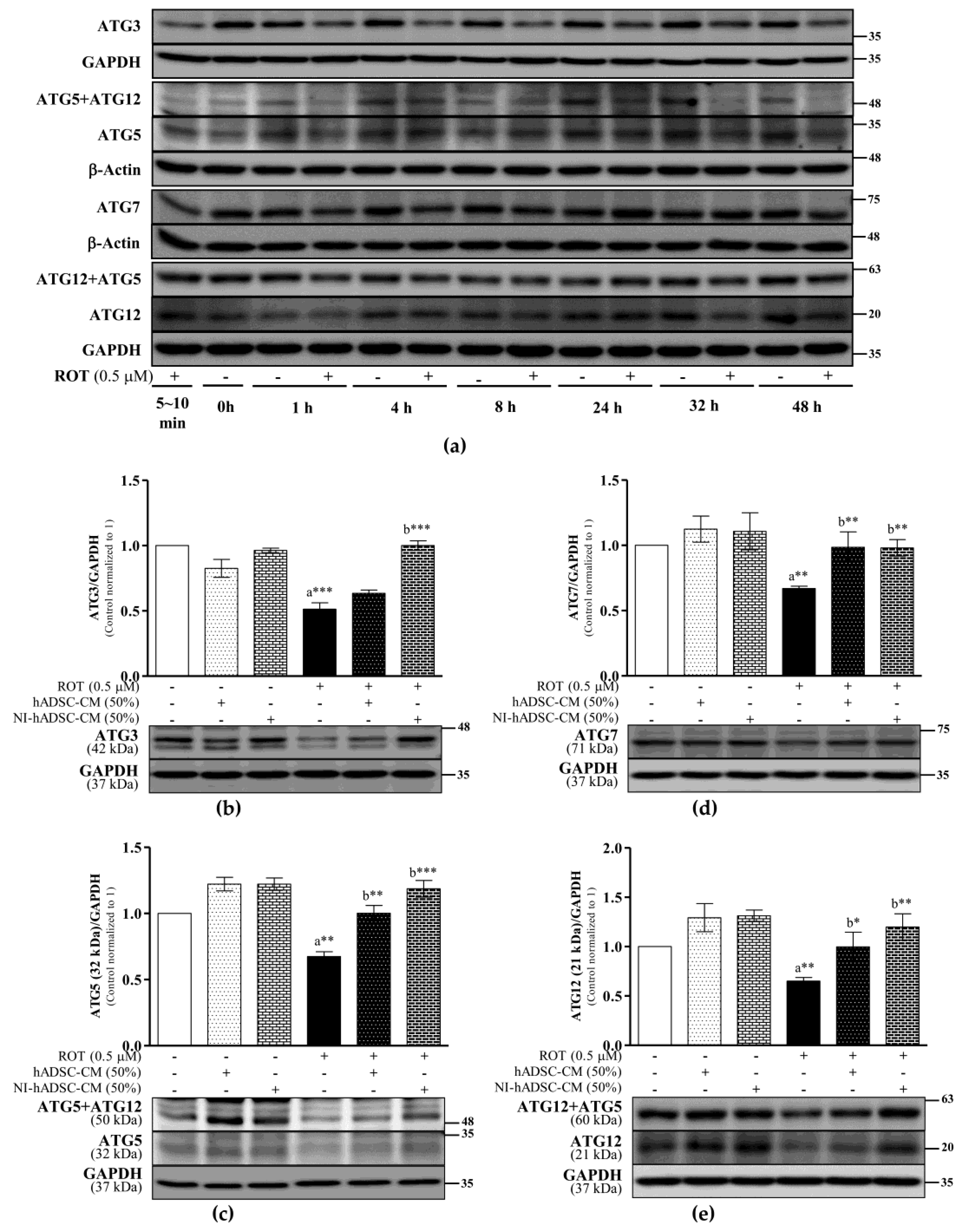
2.6. Effects of NI-hADSC-CM on ATG Protein Expression in ROT-Exposed SH-SY5Y Cells
2.7. Effects of NI-hADSC-CM on Lysosomal Protein Expression in ROT-Exposed SH-SY5Y Cells
2.8. Analysis of Autophagy Antibody Array in Conditioned Mediums
3. Discussion
4. Materials and Methods
4.1. Conditioned Medium Collection from hADSCs and NI-hADSCs
4.2. SH-SY5Y Cell Culture
4.3. Rotenone Preparation
4.4. ROT Toxicity and hADSC-CM and NI-hADSC-CM Treatments
4.5. Reactive Oxygen Species Estimation
4.6. Preparation of Triton X-100-Soluble and -Insoluble Fractions and Western Blotting of Target Proteins
4.7. Secretome Analysis by Autophagy Array Kit
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramalingam, M.; Huh, Y.J.; Lee, Y.I. The impairments of α-synuclein and mechanistic target of rapamycin in rotenone-induced SH-SY5Y cells and mice model of Parkinson’s disease. Front. Neurosci. 2019, 13, 1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peelaerts, W.; Bousset, L.; Baekelandt, V.; Melki, R. α-synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: Similarities and differences. Cell Tissue Res. 2018, 373, 195–212. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, M.; Jang, S.; Jeong, H.S. Neural-induced human adipose tissue-derived stem cells conditioned medium ameliorates rotenone-induced toxicity in SH-SY5Y cells. Int. J. Mol. Sci. 2021, 22, 2322. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Rideout, H.J.; Stefanis, L. Pathological roles of α-synuclein in neurological disorders. Lancet. Neurol. 2011, 10, 1015–1025. [Google Scholar] [CrossRef]
- Morrison Joly, M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Estrada, M.V.; Young, C.; Williams, M.; Rexer, B.N.; Sarbassov dos, D.; Muller, W.J.; et al. Rictor/mTORC2 drives progression and therapeutic resistance of HER2-amplified breast cancers. Cancer Res. 2016, 76, 4752–4764. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.G.; Lee, S.; Kim, Y.; Park, J.; Woo, D.; Kim, D.; Li, Y.; Shin, W.; Kang, H.; Yook, C.; et al. Tanc2-mediated mTOR inhibition balances mTORC1/2 signaling in the developing mouse brain and human neurons. Nat. Commun. 2021, 12, 2695. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.; Song, J.; Lu, J.; et al. Balancing mTOR signaling and autophagy in the treatment of Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschlager, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Orike, N.; Middleton, G.; Borthwick, E.; Buchman, V.; Cowen, T.; Davies, A.M. Role of PI3-kinase, Akt and Bcl-2-related proteins in sustaining the survival of neurotrophic factor-independent adult sympathetic neurons. J. Cell Biol. 2001, 154, 995–1005. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted roles of GSK-3 in cancer and autophagy-related diseases. Oxidative Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef] [Green Version]
- Jope, R.S.; Johnson, G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Ding, Q.; Xia, W.; Liu, J.C.; Yang, J.Y.; Lee, D.F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Odagiri, S.; Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in α-synucleinopathy. Acta Neuropathol. 2012, 124, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Tanji, K.; Mori, F.; Utsumi, J.; Sasaki, H.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of upstream autophagy-related proteins (ULK1, ULK2, Beclin1, VPS34 and AMBRA1) in Lewy body disease. Brain Pathol. 2016, 26, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Cheng, M.H.; Kim, S.J. Insulin suppresses MPP+-induced neurotoxicity by targeting integrins and syndecans in C6 astrocytes. J. Recept. Signal Transduct. 2017, 37, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Garrido, J.; Sancho-Shimizu, V.; Shenoy, A.R. Regulated proteolysis of p62/SQSTM1 enables differential control of autophagy and nutrient sensing. Sci. Signal. 2018, 11, eaat6903. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Kim, H.W.; Youdim, M.B.; Rhyu, I.J.; Choe, K.M.; Oh, Y.J. Binding preference of p62 towards LC3-ll during dopaminergic neurotoxin-induced impairment of autophagic flux. Autophagy 2011, 7, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [Green Version]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nature neuroscience 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Pires, A.O.; Teixeira, F.G.; Mendes-Pinheiro, B.; Serra, S.C.; Sousa, N.; Salgado, A.J. Old and new challenges in Parkinson’s disease therapeutics. Prog. Neurobiol. 2017, 156, 69–89. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Carvalho, M.M.; Panchalingam, K.M.; Rodrigues, A.J.; Mendes-Pinheiro, B.; Anjo, S.; Manadas, B.; Behie, L.A.; Sousa, N.; Salgado, A.J. Impact of the secretome of human mesenchymal stem cells on brain structure and animal behavior in a rat model of Parkinson’s disease. Stem Cells Transl. Med. 2017, 6, 634–646. [Google Scholar] [CrossRef]
- Jang, S.; Cho, H.H.; Cho, Y.B.; Park, J.S.; Jeong, H.S. Functional neural differentiation of human adipose tissue-derived stem cells using bFGF and forskolin. BMC Cell Biol. 2010, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Reviews. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009, 273, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Lamonaca, G.; Volta, M. Alpha-synuclein and LRRK2 in synaptic autophagy: Linking early dysfunction to late-stage pathology in Parkinson’s disease. Cells 2020, 9, 1115. [Google Scholar] [CrossRef]
- Jang, S.; Cho, H.H.; Kim, S.H.; Lee, K.H.; Cho, Y.B.; Park, J.S.; Jeong, H.S. Transplantation of human adipose tissue-derived stem cells for repair of injured spiral ganglion neurons in deaf guinea pigs. Neural Regen. Res. 2016, 11, 994–1000. [Google Scholar] [CrossRef]
- Jang, S.; Park, J.S.; Jeong, H.S. Neural differentiation of human adipose tissue-derived stem cells involves activation of the Wnt5a/JNK signalling. Stem Cells Int. 2015, 2015, 178618. [Google Scholar] [CrossRef]
- Baglio, S.R.; Pegtel, D.M.; Baldini, N. Mesenchymal stem cell secreted vesicles provide novel opportunities in (stem) cell-free therapy. Front. Physiol. 2012, 3, 359. [Google Scholar] [CrossRef] [Green Version]
- Vizoso, F.J.; Eiro, N.; Cid, S.; Schneider, J.; Perez-Fernandez, R. Mesenchymal stem cell secretome: Toward cell-free therapeutic strategies in regenerative medicine. Int. J. Mol. Sci. 2017, 18, 1852. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, M.; Jang, S.; Jeong, H.S. Therapeutic effects of conditioned medium of neural differentiated human bone marrow-derived stem cells on rotenone-induced α-synuclein aggregation and apoptosis. Stem Cells Int. 2021, 2021, 6658271. [Google Scholar] [CrossRef]
- Zhang, S.; Ma, Y.; Feng, J. Neuroprotective mechanisms of epsilon-viniferin in a rotenone-induced cell model of Parkinson’s disease: Significance of SIRT3-mediated FOXO3 deacetylation. Neural Regen. Res. 2020, 15, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.Y.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J. Bioenerg. Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, G.; Paciotti, S.; Gatticchi, L.; Parnetti, L. The vicious cycle between α-synuclein aggregation and autophagic-lysosomal dysfunction. Mov. Disord. 2020, 35, 34–44. [Google Scholar] [CrossRef]
- Wang, H.; Dou, S.; Zhu, J.; Shao, Z.; Wang, C.; Xu, X.; Cheng, B. Ghrelin protects against rotenone-induced cytotoxicity: Involvement of mitophagy and the AMPK/SIRT1/PGC1α pathway. Neuropeptides 2021, 87, 102134. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [Green Version]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of α-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.P.; Hewitt, E.W. Cellular proteostasis: Degradation of misfolded proteins by lysosomes. Essays Biochem. 2016, 60, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Sun, X.; Wang, K.; Yu, Y.; Zhang, L.; Zhang, K.; Gu, J.; Yuan, X.; Song, G. Hydrogen-saturated saline mediated neuroprotection through autophagy via PI3K/AKT/mTOR pathway in early and medium stages of rotenone-induced Parkinson’s disease rats. Brain Res. Bull. 2021, 172, 1–13. [Google Scholar] [CrossRef]
- Yin, S.; Liu, L.; Gan, W. The roles of post-translational modifications on mTOR signaling. Int. J. Mol. Sci. 2021, 22, 1784. [Google Scholar] [CrossRef]
- Zhou, Q.; Liu, C.; Liu, W.; Zhang, H.; Zhang, R.; Liu, J.; Zhang, J.; Xu, C.; Liu, L.; Huang, S.; et al. Rotenone induction of hydrogen peroxide inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E pathways, leading to neuronal apoptosis. Toxicol. Sci. 2015, 143, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Makris, A.; Nemsadze, G.; Brunt, A.M.; Kuemmel, S.; et al. Fulvestrant plus vistusertib vs fulvestrant plus everolimus vs fulvestrant alone for women with hormone receptor-positive metastatic breast cancer: The MANTA phase 2 randomized clinical trial. JAMA Oncol. 2019, 5, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Bapat, P.; Deogharkar, A.; Kazi, S.; Singh, S.K.V.; Gupta, T.; Jalali, R.; Sridhar, E.; Moiyadi, A.; Shetty, P.; et al. MiR-592 activates the mTOR kinase, ERK1/ERK2 kinase signaling and imparts neuronal differentiation signature characteristic of group 4 medulloblastoma. Hum. Mol. Genet. 2021, 30, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Enogieru, A.B.; Haylett, W.; Hiss, D.C.; Ekpo, O.E. Regulation of AKT/AMPK signaling, autophagy and mitigation of apoptosis in Rutin-pretreated SH-SY5Y cells exposed to MPP+. Metab. Brain Dis. 2021, 36, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, C.; Chen, S.; Ye, Y.; Guo, M.; Ren, Q.; Liu, L.; Zhang, H.; Xu, C.; Zhou, Q.; et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell. Signal. 2014, 26, 1680–1689. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Bouhamida, E.; Danese, A.; Previati, M.; Pinton, P.; Patergnani, S. Relevance of autophagy and mitophagy dynamics and markers in neurodegenerative diseases. Biomedicines 2021, 9, 149. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [CrossRef]
- Ramalingam, M.; Kim, S.J. Protective effects of activated signaling pathways by insulin on C6 glial cell model of MPP+-induced Parkinson’s disease. J. Recept. Signal Transduct. 2017, 37, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Kim, S.J. The neuroprotective role of insulin against MPP+-induced Parkinson’s disease in differentiated SH-SY5Y cells. J. Cell. Biochem. 2016, 117, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampada, A.; O’Prey, J.; Szabadkai, G.; Ryan, K.M.; Hochhauser, D.; Salomoni, P. mTORC1-independent autophagy regulates receptor tyrosine kinase phosphorylation in colorectal cancer cells via an mTORC2-mediated mechanism. Cell Death Differ. 2017, 24, 1045–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Reviews. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Chen, G.; Fan, Z.; Luo, J.; Ke, Z.J. GSK3beta and endoplasmic reticulum stress mediate rotenone-induced death of SK-N-MC neuroblastoma cells. Biochem. Pharmacol. 2008, 76, 128–138. [Google Scholar] [CrossRef]
- Wills, J.; Credle, J.; Haggerty, T.; Lee, J.H.; Oaks, A.W.; Sidhu, A. Tauopathic changes in the striatum of A53T α-synuclein mutant mouse model of Parkinson’s disease. PLoS ONE 2011, 6, e17953. [Google Scholar] [CrossRef] [Green Version]
- Credle, J.J.; George, J.L.; Wills, J.; Duka, V.; Shah, K.; Lee, Y.C.; Rodriguez, O.; Simkins, T.; Winter, M.; Moechars, D.; et al. GSK-3beta dysregulation contributes to parkinson’s-like pathophysiology with associated region-specific phosphorylation and accumulation of tau and α-synuclein. Cell Death Differ. 2015, 22, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Liu, L.; Li, C.J.; Lu, Y.; Zong, X.G.; Luo, C.; Sun, J.; Guo, L.J. Baclofen mediates neuroprotection on hippocampal CA1 pyramidal cells through the regulation of autophagy under chronic cerebral hypoperfusion. Sci. Rep. 2015, 5, 14474. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; He, X.; Hsu, J.M.; Xia, W.; Chen, C.T.; Li, L.Y.; Lee, D.F.; Liu, J.C.; Zhong, Q.; Wang, X.; et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol. 2007, 27, 4006–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reszka, A.A.; Seger, R.; Diltz, C.D.; Krebs, E.G.; Fischer, E.H. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc. Natl. Acad. Sci. USA 1995, 92, 8881–8885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweatt, J.D. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 2004, 14, 311–317. [Google Scholar] [CrossRef]
- Langlais, P.; Yi, Z.; Mandarino, L.J. The identification of raptor as a substrate for p44/42 MAPK. Endocrinology 2011, 152, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Theus, M.H.; Wei, L. Role of ERK 1/2 signaling in neuronal differentiation of cultured embryonic stem cells. Dev. Growth Differ. 2006, 48, 513–523. [Google Scholar] [CrossRef]
- Bruneau, S.; Nakayama, H.; Woda, C.B.; Flynn, E.A.; Briscoe, D.M. DEPTOR regulates vascular endothelial cell activation and proinflammatory and angiogenic responses. Blood 2013, 122, 1833–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Klionsky, D.J. Physiological functions of Atg6/Beclin 1: A unique autophagy-related protein. Cell Res. 2007, 17, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.A.; Gao, F.B. Regulation of Abeta pathology by beclin 1: A protective role for autophagy? J. Clin. Investig. 2008, 118, 2015–2018. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, K.J.; Ademi, C.; Bertram, S.; Schmid, K.W.; Baba, H.A. Prognostic relevance of autophagy-related markers LC3, p62/sequestosome 1, Beclin-1 and ULK1 in colorectal cancer patients with respect to KRAS mutational status. World J. Surg. Oncol. 2016, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, J.; Credle, J.; Oaks, A.W.; Duka, V.; Lee, J.H.; Jones, J.; Sidhu, A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS ONE 2012, 7, e30745. [Google Scholar] [CrossRef]
- Higashi, S.; Moore, D.J.; Minegishi, M.; Kasanuki, K.; Fujishiro, H.; Kabuta, T.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Localization of MAP1-LC3 in vulnerable neurons and Lewy bodies in brains of patients with dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2011, 70, 264–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Stefanis, L. α-synuclein degradation by autophagic pathways: A potential key to Parkinson’s disease pathogenesis. Autophagy 2008, 4, 917–919. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.S.; Ho, C.S.; Huang, Y.W.; Wu, T.Y.; Lee, T.H.; Huang, Z.D.; Wang, T.J.; Yang, S.J.; Chiang, M.F. Impairment of proteasome and autophagy underlying the pathogenesis of Leukodystrophy. Cells 2020, 9, 1124. [Google Scholar] [CrossRef]
- Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol. Dis. 2011, 43, 690–697. [Google Scholar] [CrossRef]
- Cho, J.H.; Kim, K.; Kim, S.A.; Park, S.; Park, B.O.; Kim, J.H.; Kim, S.Y.; Kwon, M.J.; Han, M.H.; Lee, S.B.; et al. Deubiquitinase OTUD5 is a positive regulator of mTORC1 and mTORC2 signaling pathways. Cell Death Differ. 2021, 28, 900–914. [Google Scholar] [CrossRef]
- Joung, I.; Kim, H.J.; Kwon, Y.K. p62 modulates Akt activity via association with PKCzeta in neuronal survival and differentiation. Biochem. Biophys. Res. Commun. 2005, 334, 654–660. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Wang, Y.; Ballar, P.; Zhong, Y.; Zhang, X.; Liu, C.; Zhang, Y.J.; Monteiro, M.J.; Li, J.; Fang, S. SVIP induces localization of p97/VCP to the plasma and lysosomal membranes and regulates autophagy. PLoS ONE 2011, 6, e24478. [Google Scholar] [CrossRef]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suaga, P.; Luzon-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.J.; Feng, Y.; Liu, T.T.; Liu, X.; Bao, J.J.; Shi, A.M.; Hu, D.M.; Liu, T.; Yu, Y.L. Thioredoxin-interacting protein induced α-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 2017, 23, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to α-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.C.; Erustes, A.G.; Reckziegel, P.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. α-Synuclein overexpression induces lysosomal dysfunction and autophagy impairment in human neuroblastoma SH-SY5Y. Neurochem. Res. 2020, 45, 2749–2761. [Google Scholar] [CrossRef]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [Green Version]
- Usategui-Martin, R.; Puertas-Neyra, K.; Garcia-Gutierrez, M.T.; Fuentes, M.; Pastor, J.C.; Fernandez-Bueno, I. Human mesenchymal stem cell secretome exhibits a neuroprotective effect over in vitro retinal photoreceptor degeneration. Mol. Ther. Methods Clin. Dev. 2020, 17, 1155–1166. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramalingam, M.; Jeong, H.-S.; Hwang, J.; Cho, H.-H.; Kim, B.C.; Kim, E.; Jang, S. Autophagy Signaling by Neural-Induced Human Adipose Tissue-Derived Stem Cell-Conditioned Medium during Rotenone-Induced Toxicity in SH-SY5Y Cells. Int. J. Mol. Sci. 2022, 23, 4193. https://doi.org/10.3390/ijms23084193
Ramalingam M, Jeong H-S, Hwang J, Cho H-H, Kim BC, Kim E, Jang S. Autophagy Signaling by Neural-Induced Human Adipose Tissue-Derived Stem Cell-Conditioned Medium during Rotenone-Induced Toxicity in SH-SY5Y Cells. International Journal of Molecular Sciences. 2022; 23(8):4193. https://doi.org/10.3390/ijms23084193
Chicago/Turabian StyleRamalingam, Mahesh, Han-Seong Jeong, Jinsu Hwang, Hyong-Ho Cho, Byeong C. Kim, Eungpil Kim, and Sujeong Jang. 2022. "Autophagy Signaling by Neural-Induced Human Adipose Tissue-Derived Stem Cell-Conditioned Medium during Rotenone-Induced Toxicity in SH-SY5Y Cells" International Journal of Molecular Sciences 23, no. 8: 4193. https://doi.org/10.3390/ijms23084193