1. Introduction
Dry eye disease (DED), as defined in the recent Dry Eye Workshop II (DEWS II) report, “is a multifactorial disease of the ocular surface characterized by a loss of homeostasis of the tear film, accompanied by ocular symptoms, in which tear film instability and hyperosmolarity, ocular surface inflammation and damage, and neurosensory abnormalities play etiological roles” [
1]. This chronic and often painful disease affects an estimated 16.4 million Americans alone, increases in prevalence with age, and disproportionately affects females more than males [
2,
3]. DED is the leading reason that people in the United States seek help from an eye care physician [
4]. DED is associated with a significant financial, psychological, and emotional burden due to time lost from work, the high cost of care, chronic pain, and in advanced disease, depression and disability. Moreover, the impact of DED in patients’ day-to-day lives is shown to be comparable to that of moderate-to-severe angina [
5]. With no effective treatments or cure, DED creates an enormous public health burden and substantially decreases the overall quality of life in those afflicted.
The underlying pathobiology of DED is linked to hyperosmolarity of the precorneal tear film in both major subtypes: aqueous-deficient DED and evaporative DED [
6,
7,
8]. In aqueous-deficient DED, the lacrimal gland is unable to produce enough aqueous to adequately bathe the surface of the corneal and conjunctival epithelium [
6]. In evaporative DED, there is often sufficient aqueous production. Instead, tear film evaporation occurs due to a reduction and alteration in the composition of lipids being produced and released from the meibomian glands that reside in the upper and lower eyelids [
6,
9]. Both aqueous-deficient and evaporative DED result in an increase in tear osmolarity. This triggers a complex chain of inflammatory events that includes the activation of Toll-like receptors and mitogen-activated protein kinase (MAPK) pathways [
10,
11,
12,
13,
14,
15,
16,
17]. Dysfunctional aquaporin 5 (AQP5) can also impair water channels present in corneal epithelial cells, leading to the activation of c-Jun N-terminal kinase (JNK1/2) MAPK and an increase in proinflammatory cytokine expression [
18]. Similarly, the transient receptor potential vanilloid channel type 1 (TRPV1) signaling pathway mediates the release of inflammatory cytokines (IL-6 and IL-8) through EGFR transactivation, MAPK, and NF-κB stimulation in corneal epithelial cells exposed to hyperosmolar stress [
19]. Together, these hyperosmolar induced cascades of pro-inflammatory cytokines and chemokines, along with matrix metalloproteinases that degrade tissue, result in increased ocular surface damage [
20,
21,
22,
23,
24,
25,
26,
27].
The ocular surface is formed by two contiguous epithelia: the corneal and conjunctival epithelium. In recent years, hyperosmolarity has been linked to a variety of mitochondrial changes in both corneal and conjunctival epithelial cells, including oxidative stress, mitochondrial DNA damage and apoptosis [
23,
24,
28,
29,
30]. This hyperosmolar-mediated increase in apoptosis is associated with a corresponding loss of barrier function, which is essential to protect the cornea from environmental insults, sheer stress, and pathogen invasion [
29,
31]. Hyperosmolarity has also been shown to increase oxidative stress and exert negative effects on mitochondria in other non-ocular epithelial cells, such as intestinal and bronchial epithelia [
32,
33,
34,
35,
36,
37]. This includes the upregulation of proinflammatory mediators in these cell types. Thus, in addition to ocular surface disease, hyperosmolar stress is a major underlying factor in the pathophysiology of many inflammatory lung and bowel disorders.
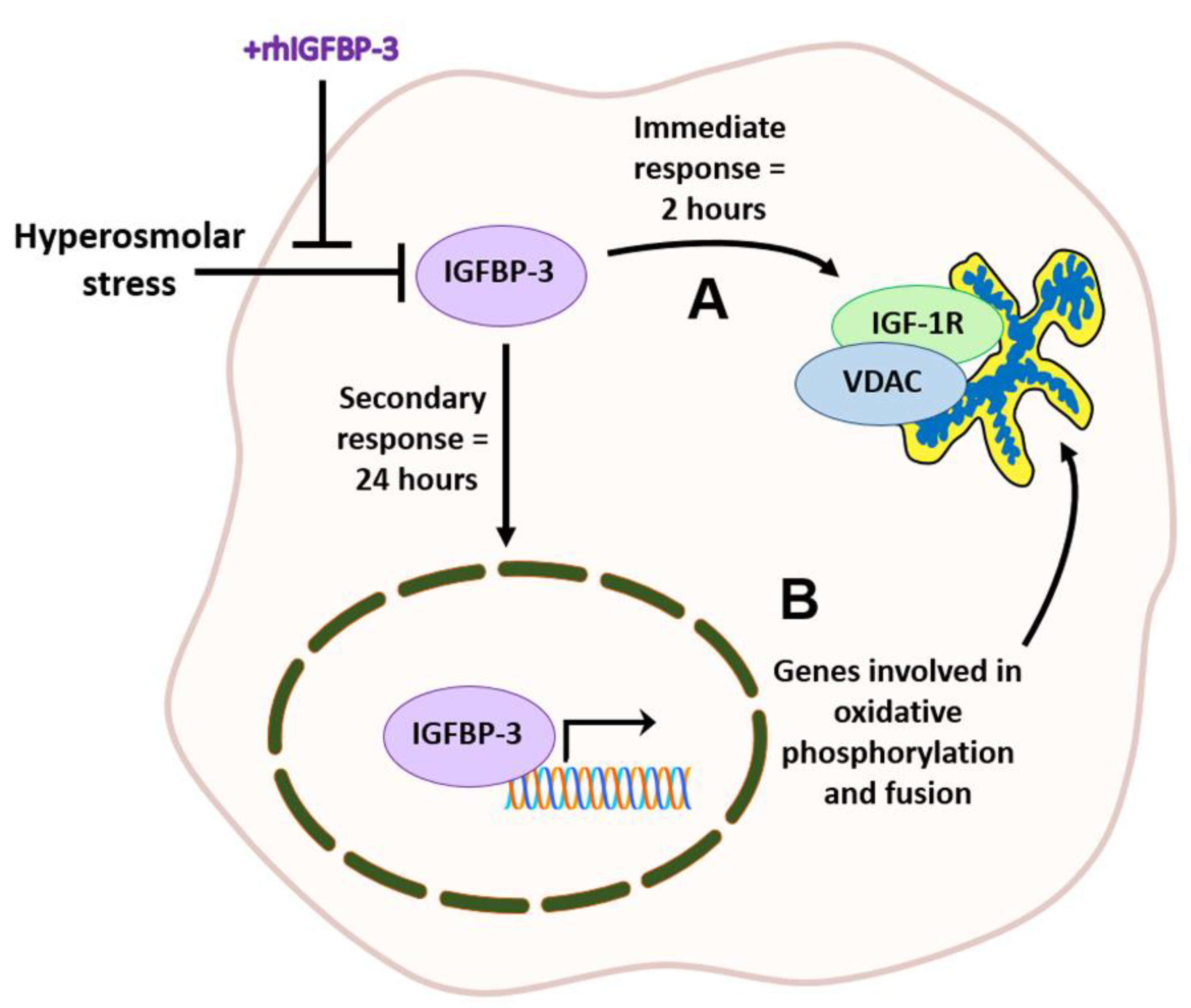
The insulin-like growth factor (IGF) family consists of ligands, cell surface receptors, and insulin-like binding proteins (IGFBPs) that function to mediate cell growth and survival throughout the body. Of the six known binding proteins, IGFBP-3 is the most abundant binding protein in circulation where it functions to block IGF-1-mediated activation of the type 1 IGF receptor [
38]. IGFBP-3 is also produced locally in many tissues and is present within the human precorneal tear film [
39,
40]. We have previously shown that IGFBP-3 is downregulated in response to hyperosmolar stress in corneal epithelial cells [
41]. We further identified a novel role for IGFPB-3 in mitochondrial homeostasis and quality control in corneal and conjunctival epithelial cells [
42]. These intricate networks are essential for optimal mitochondrial function and epithelial cell health. In contrast, mitochondrial failure or dysfunction triggers degenerative cellular processes, such as those seen in DED. In this study, we interrogated the role of IGFBP-3 in mediating mitochondrial morphology and metabolic activity in corneal and conjunctival epithelial cells exposed to hyperosmolar stress. Using a mouse model of aqueous-deficient DED, we further investigated the effect of topical treatment with exogenous IGFBP-3 on the ocular surface in vivo.
3. Discussion
We recently reported that IGFBP-3 functions to inhibit mitophagy and promote mitochondrial respiration in corneal and conjunctival epithelial cells [
42]. We have further shown that IGFBP-3 is decreased during hyperosmolar stress, and thus may play a role in stabilizing mitochondria [
41]. The mechanism by which this occurs is unclear. In the present study, we sought to interrogate the role of IGFBP-3 in mitochondrial and metabolic activity in response to hyperosmolar stress in vitro and in a mouse model of aqueous-deficient dry eye in vivo. Importantly, we found that IGFBP-3 expression and secretion are directly related to changes in mitochondrial respiration in response to increasing osmolarity. Specifically, we found that small increases in hyperosmolarity drive an increase in IGFBP-3 expression and secretion, along with increased mitochondrial respiration. This finding is consistent with our hyperglycemia model where cultures in elevated glucose also led to an increase in IGFBP-3 [
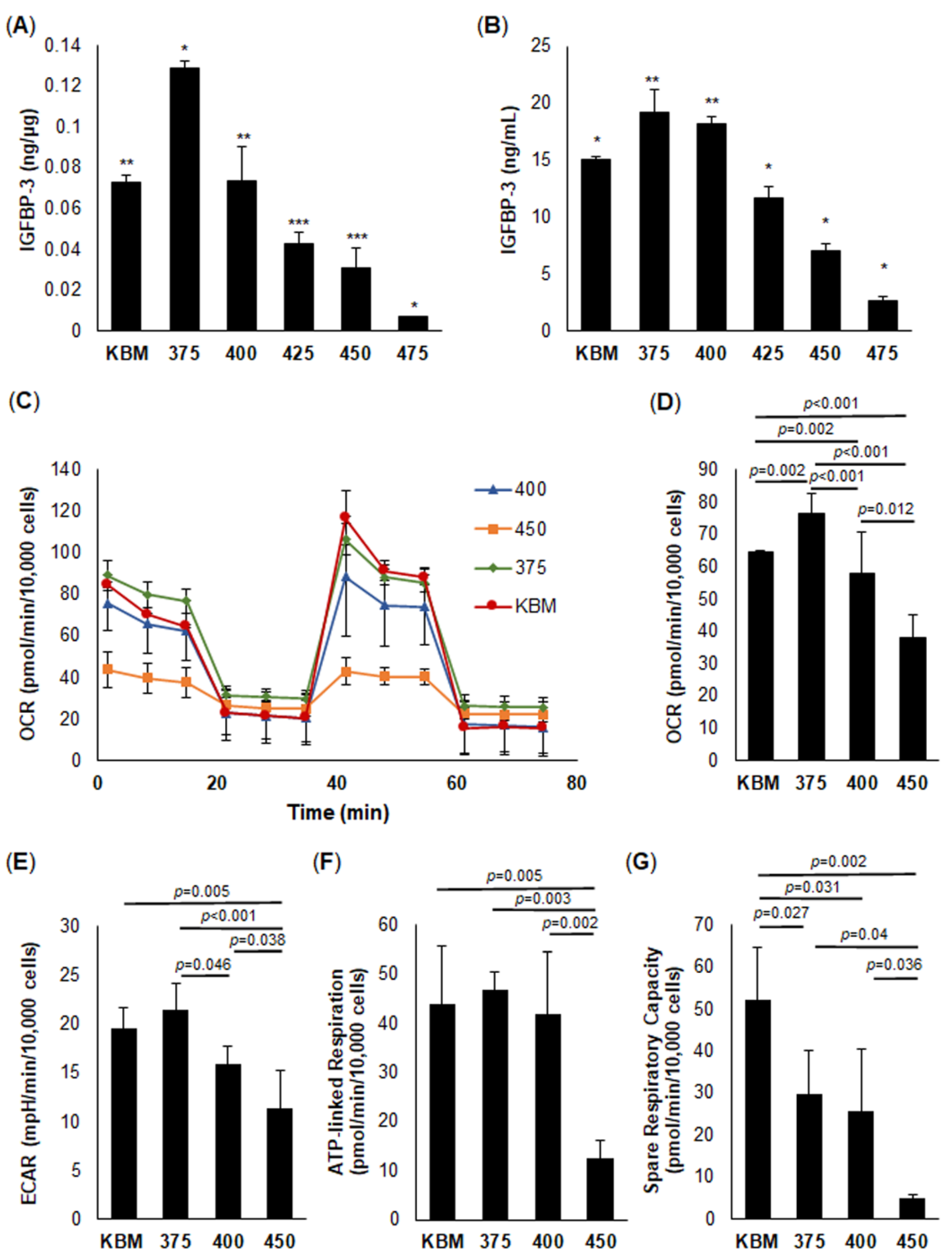
39]. Interestingly, however, as osmolarity continued to increase, IGFBP-3 levels decreased in a concentration-dependent manner. This was reflected by a parallel decrease in respiration.
In contrast to respiration, spare respiratory capacity was decreased compared to the isotonic condition at all concentrations tested. Since spare respiratory capacity is a measure of a cell’s energy reserves, even low levels of hyperosmolarity are sufficient to disrupt the capacity of a cell to undergo metabolic adaptation and survival [
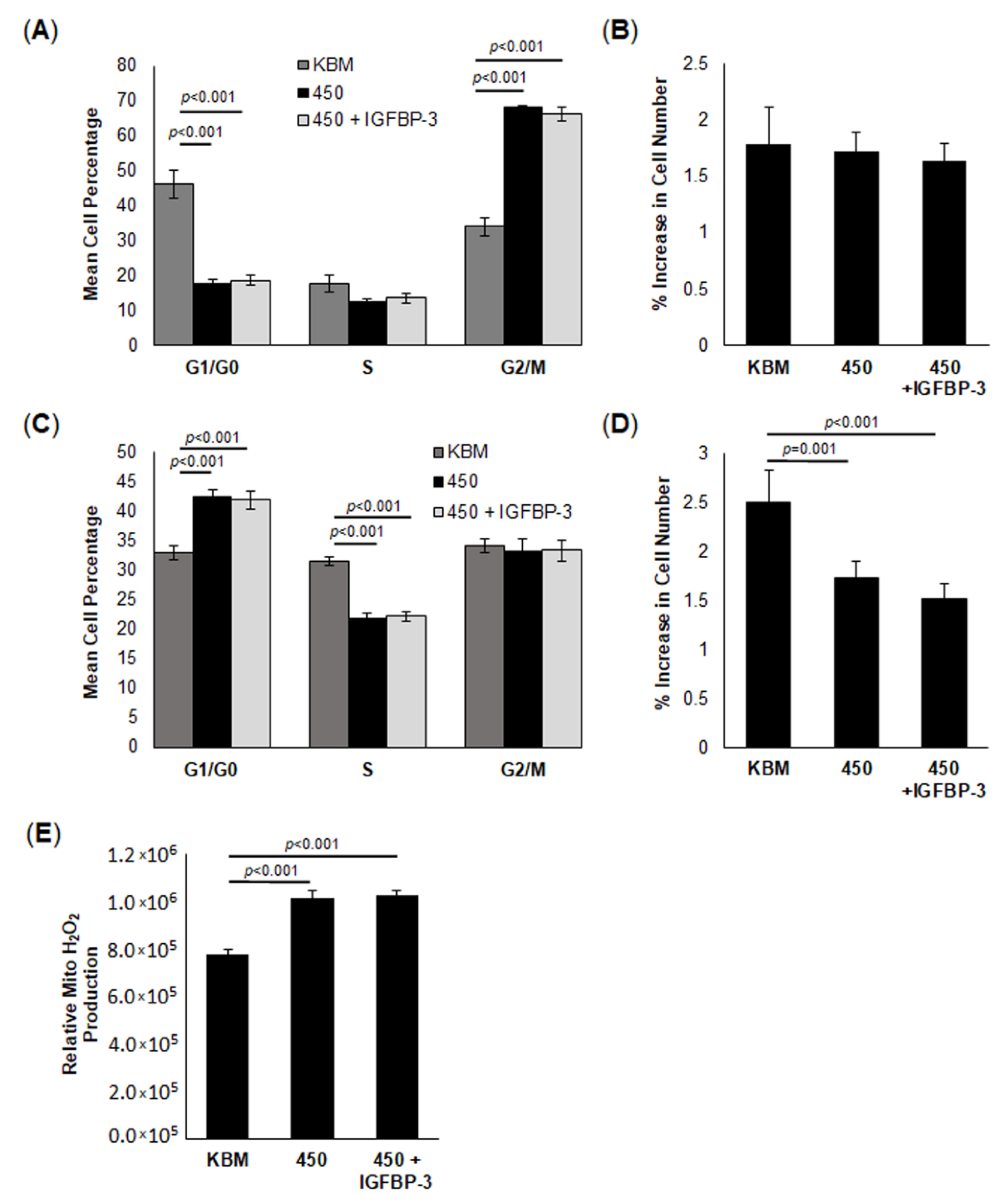
45]. We speculate that the increase in IGFBP-3 at the lowest osmolar stress tested (375 mOsm) was due to the cell trying to draw on their reserve to meet an increased demand for energy. As osmolarity continued to increase, there was an inhibition of this adaptive mechanism. This, coupled with the reported loss of IGFBP-3 with increasing osmolarity, suggests that IGFBP-3 is an important mediator of corneal epithelial cell stress responses whereby the increase in IGFBP-3 in response to mild or moderate stress is protective. This protective effect is lost in the presence of a substantial stress that downregulates IGFBP-3, leading to a loss of cell viability. Moreover, the measured changes in IGFBP-3 levels were not restricted to corneal epithelial cells, but were evident in conjunctival cells, which are also subject to chronic hyperosmolar stress in dry eye disease.
A surprising feature of this model was the corresponding decrease in the extracellular acidification rate, a surrogate marker for glycolysis. We have previously shown that growth factor deprivation decreases glycolysis and that this is associated an increase in respiration [
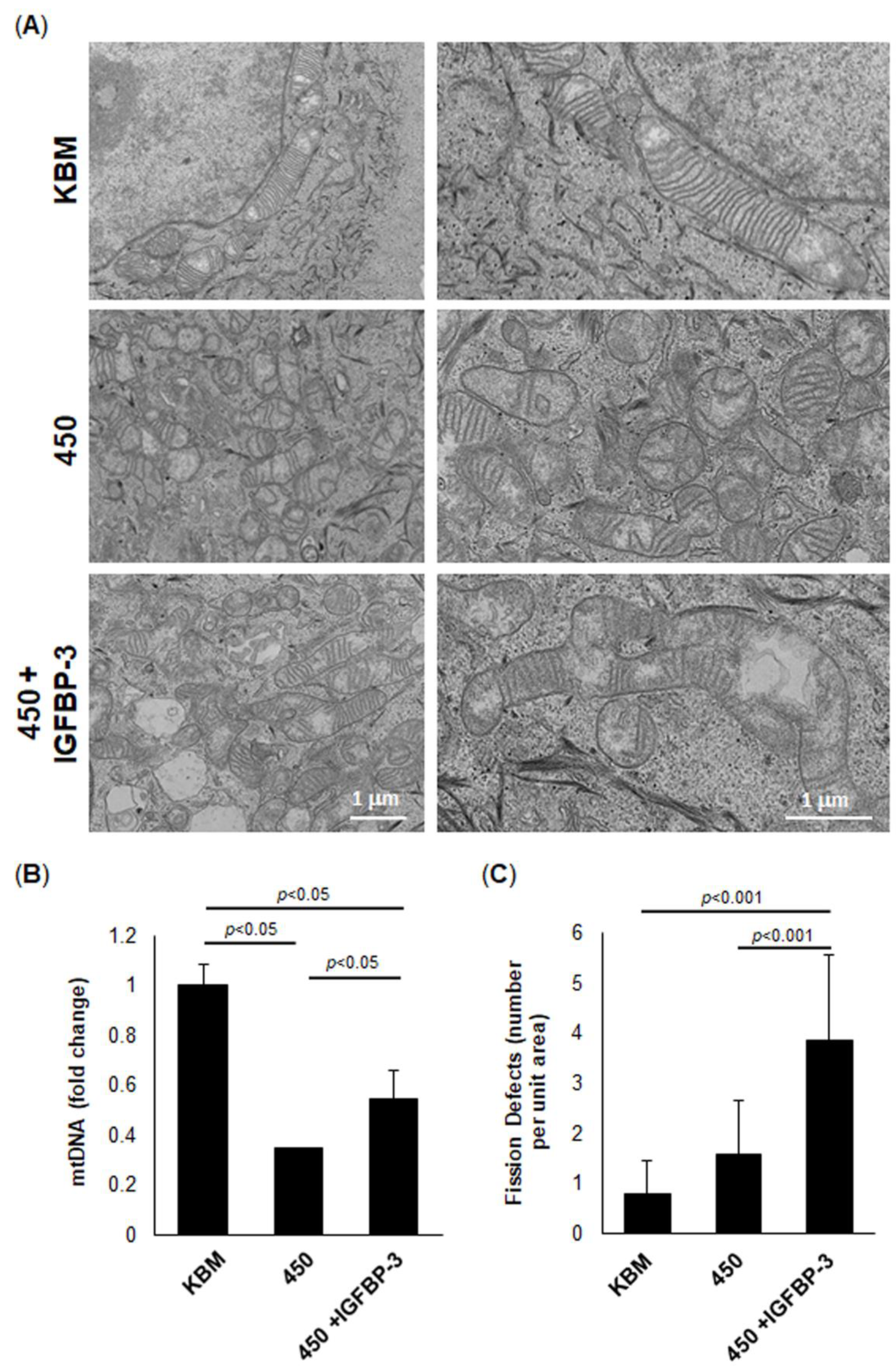
43]. However, if the stress is prolonged until mitochondrial respiration decreases due to robust depolarization, glycolysis increases to sustain the cell. Given the high levels of glycogen in the corneal epithelium, the increase in glycolysis is likely due to enhanced glycogenolysis. In contrast to this, we found that hyperosmolar stress decreased both respiration and glycolysis. This occurred in concert with a reduction in protein translation. Thus, in the absence of IGFBP-3, both glycolysis and respiration are suppressed. This was associated with robust mitochondrial fission, as seen by electron microscopy, and the loss of mtDNA. Since fission is essential for mitophagy to occur and attempt to repair damaged mitochondria, these findings would suggest an induction of mitophagy [
46]. This is supported by our prior report showing that the loss of IGFBP-3 promotes mitophagy in these same epithelial cells [
42]. An unchecked increase in mitophagy can lead to apoptotic cell death, the latter of which has been shown by other studies to occur in response to hyperosmolar stress and in DED [
47].
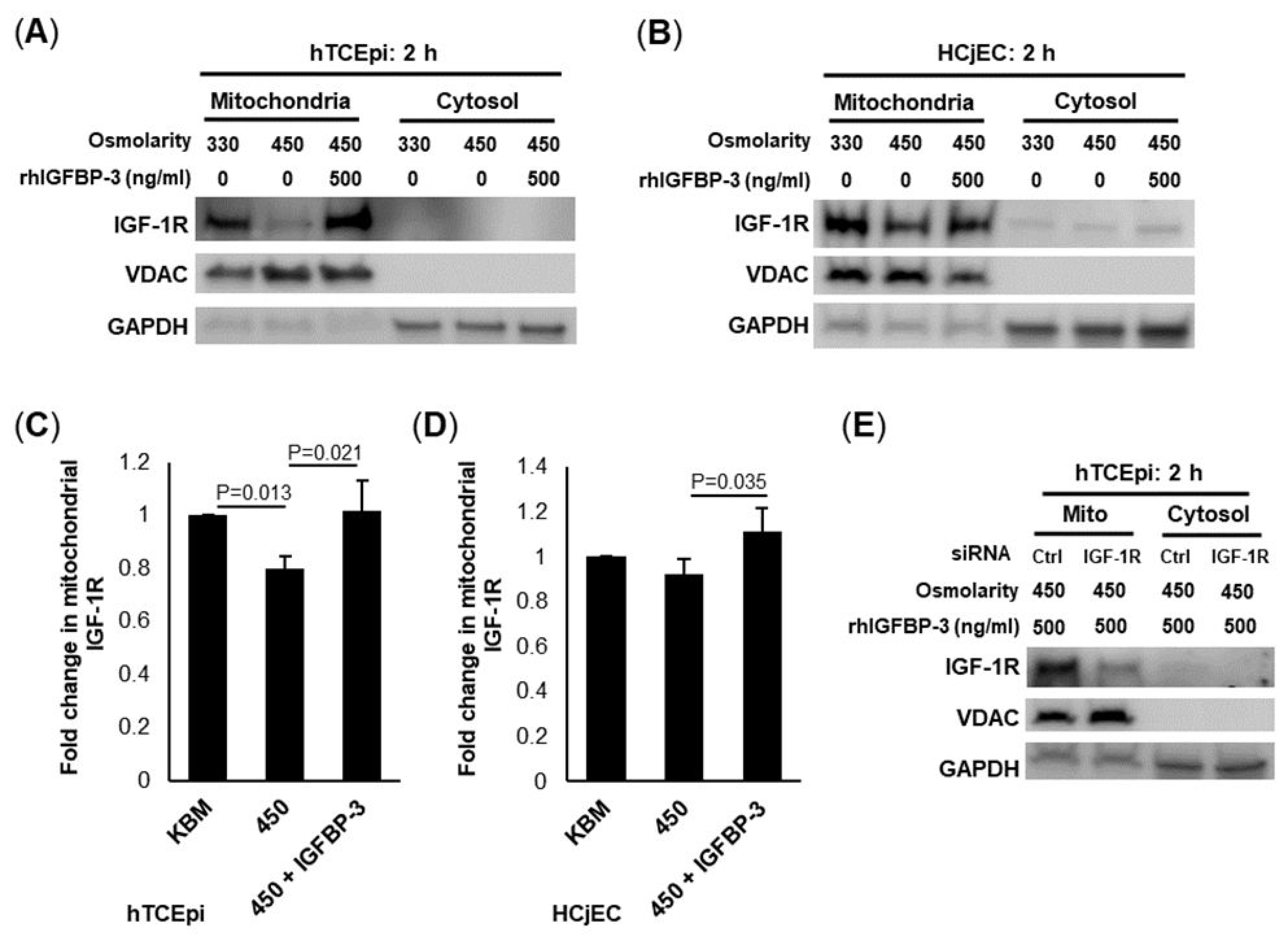
The principal function for IGFBP-3 is to bind IGF-1, inhibiting the IGF-1-mediated activation of IGF-1R. In addition to these IGF-1-dependent effects, IGFBP-3 has also been shown to have IGF-1-independent functions. In the corneal epithelium, we have previously reported that IGFBP-3 mediates the intracellular trafficking of the IGF-1R/INSR hybrid (Hybrid-R) in the absence of IGF-1 [
44,
48,
49]. This occurs through IGFBP-3-mediated SUMOylation of IGF-1R. Here, again, we report on the capacity of IGFBP-3 to induce intracellular trafficking, our second key finding. More specifically, we show that IGFBP-3 drives the mitochondrial localization of IGF-1R. The increase in mitochondrial IGF-1R occurs early in the stress response and was readily detectable by 2 h, as opposed to later changes that we documented after 24 h of hyperosmolar stress. The finding of a mitochondrial-localized IGF-1R during hyperosmolar stress is consistent with our prior work, showing IGF-1R, INSR and EGFR all localizing to the outer mitochondrial membrane [
43].
With respect to INSR, we previously documented the novel interaction between INSR and the voltage-dependent anion channel (VDAC) [
43]. The disruption of this interaction led to mitochondrial instability, depolarization and fragmentation. Since IGFBP-3 induces the shift of IGF-1R to mitochondria during the early stages of hyperosmolar stress, we speculate that it may play an important role in mitochondrial stabilization, while the cell redistributes resources to facilitate metabolic adaptation. This finding is further supported by work showing that IGF-1R functions to maintain mitochondrial integrity and prevent apoptosis [
50]. This occurs through the canonical IGF-1/IGF-1R signaling pathway. The ability of IGF-1R to physically stabilize mitochondria and promote survival during stress reflects a previously unrecognized IGF-1-independent mechanism. Mitochondrial-localized IGF-1R was also evident in conjunctival epithelial cells, indicating this is not cornea-specific and may represent a generalized mechanism.
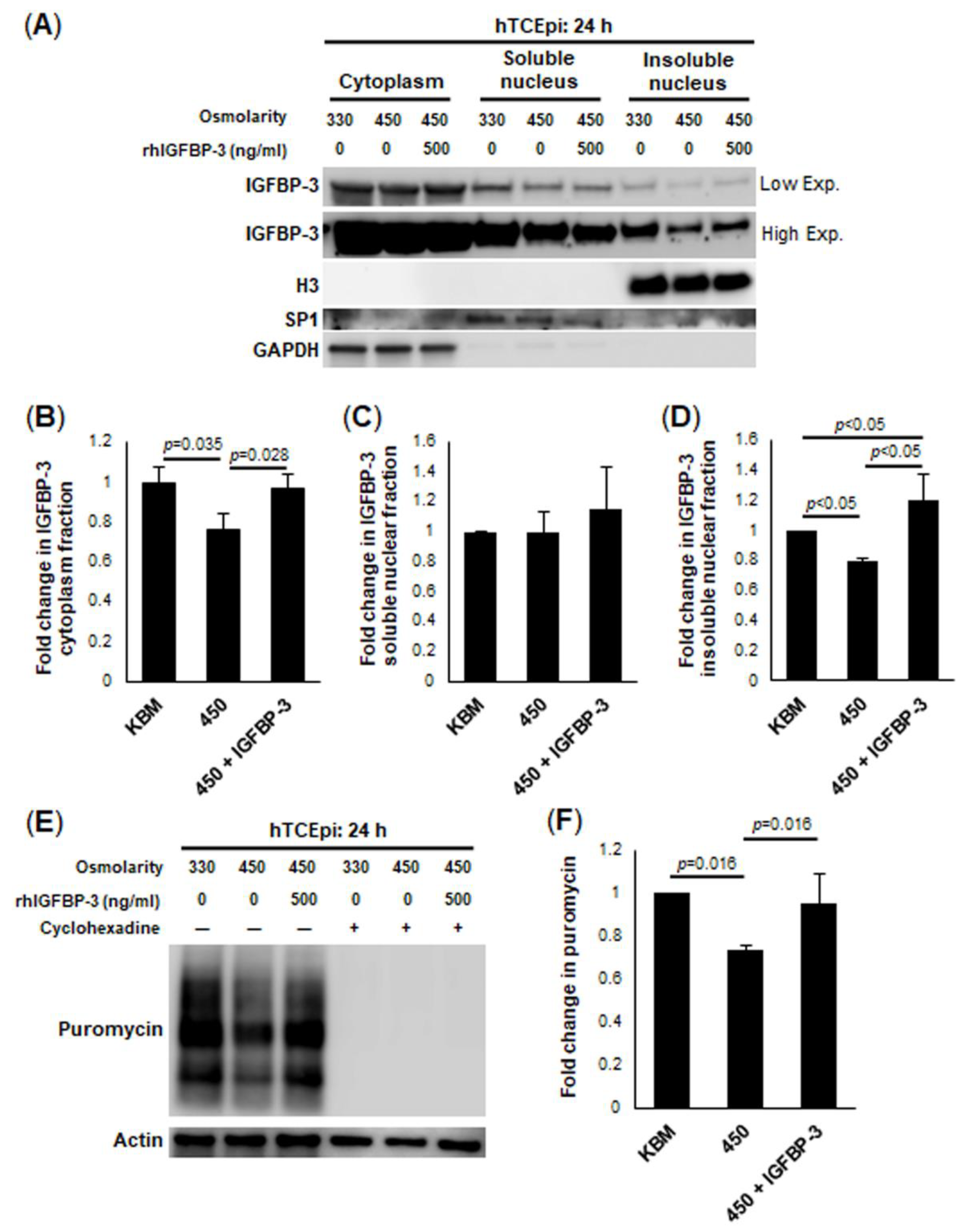
In contrast to these early changes, after 24 h of hyperosmolar stress, there was a decrease in intra- and extracellular levels of IGFBP-3. This corresponded to a decrease in puromycin-labeled proteins, confirming a hyperosmolar-mediated decrease in protein translation. Co-treatment with IGFBP-3 not only maintained protein translation at an isotonic level, but increased levels of IGFBP-3 in the insoluble nucleus. In our ongoing mouse studies, we similarly found that knockdown of IGFBP-3 using siRNA oligonucleotides administered by subconjunctival injection, also triggered the translocation of IGFBP-3 to the nucleus (unpublished data). The nuclear role of IGFBP-3 in gene transcription has been previously reported [
51,
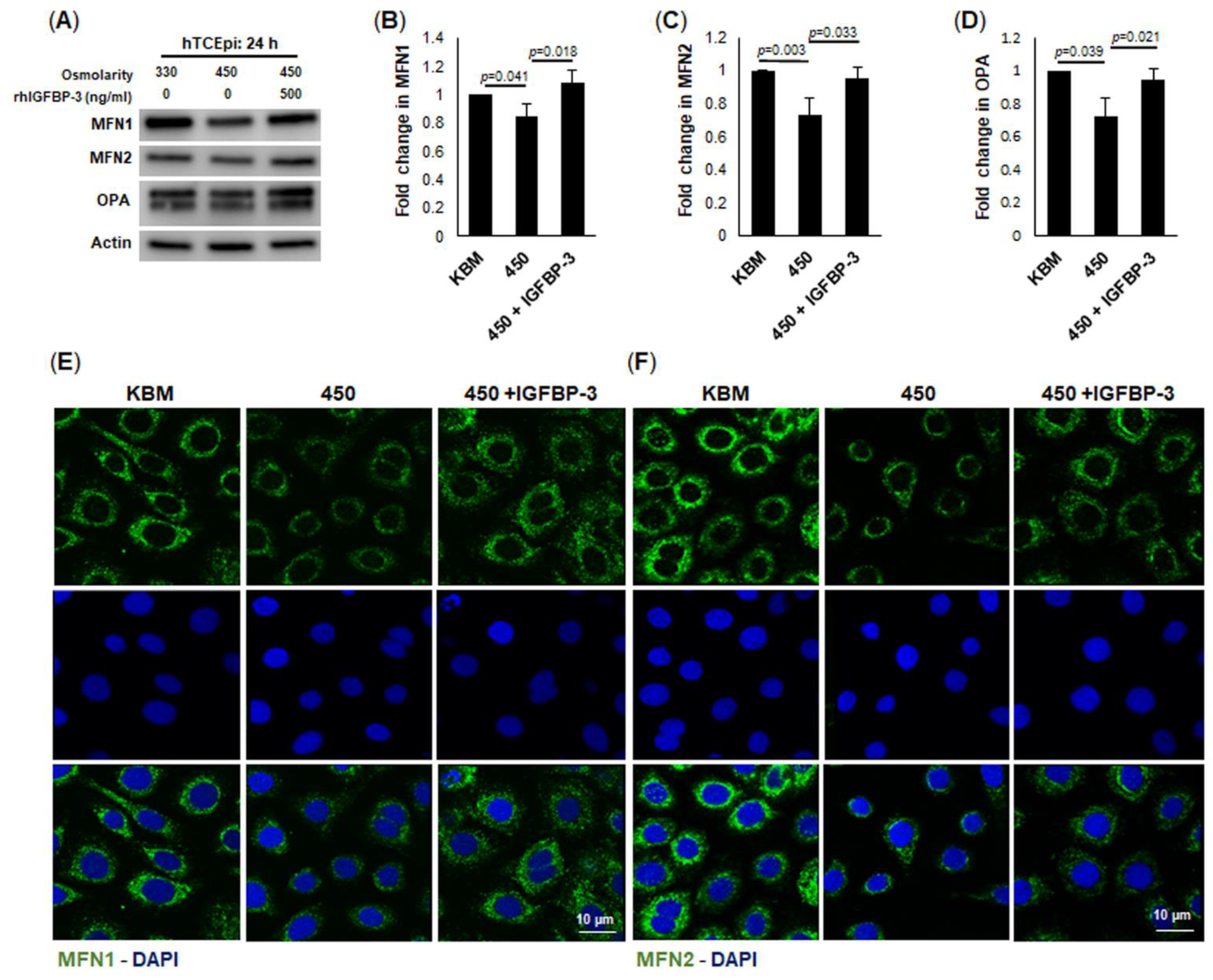
52]. Given the close relationship between IGFBP-3 expression and mitochondrial respiration, these data suggest that nuclear IGFBP-3 may promote the expression of genes essential for oxidative phosphorylation. In addition to an IGFPB-3-mediated increase in oxidative phosphorylation, we also found an increase in the outer mitochondrial fusion proteins MFN1 and MFN2, along with an increase in the inner mitochondrial fusion protein, OPA. Of note, OPA also has an integral role in cristae formation [
53]. Together, the increase in MFN1 and OPA was shown to drive stress-induced mitochondrial hyperfusion [
54]. Indeed, exogenous IGFPB-3 led to the development of hyperfused mitochondria with well-defined cristae and an attenuation of hyperosmolar-mediated loss of mtDNA.
Various types of stress have been shown to disrupt normal mitochondrial dynamics. As is the case illustrated here with hyperosmolarity, mitochondria undergo robust fission. This enables the clearance of damaged mitochondria through mitophagy and is associated with a loss of spare respiratory capacity. In contrast, mitochondria undergo transient hyperfusion in the presence of IGFBP-3. This leads to an increase in respiration and ATP generation, in order to generate the required energy needed to stabilize the cell and mediate metabolic adaptation. In our current model, exogenous IGFBP-3 blocks mitophagy through an increase in fission defects, resulting in hyperfused mitochondria. IGFBP-3 also enhances the lamellar cristae structure, setting the stage for enhanced metabolic capacity. While the mechanism(s) underlying the mitochondrial and metabolic effects of IGFBP-3 are still not well understood, these data point to the clear role of IGFBP-3 as a key mediator of epithelial cell stress responses.
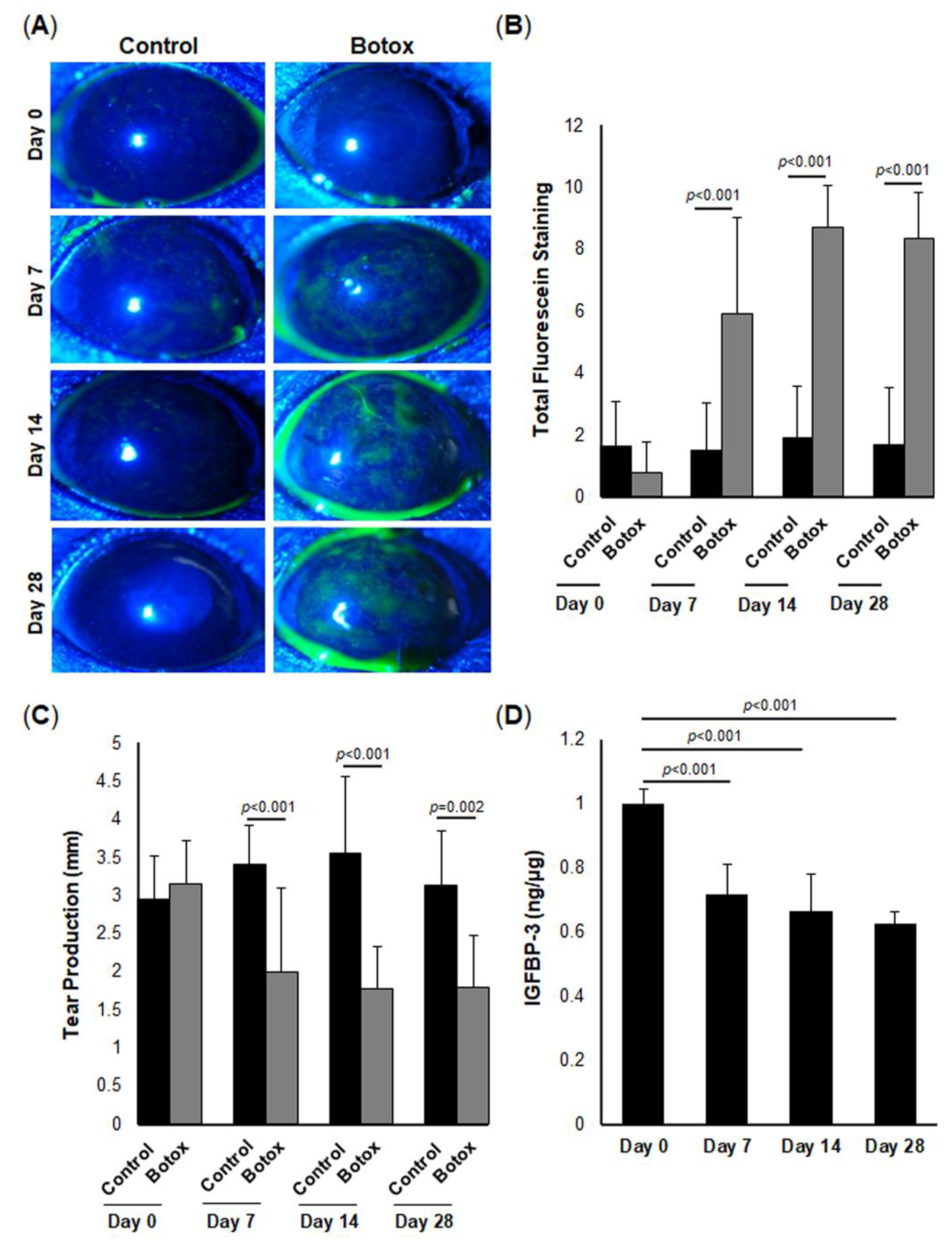
We further illustrated the physiological and translational relevance of IGFBP-3 in vivo, using an aqueous-deficient mouse model of DED. This was accomplished by injecting botulinum toxin into the exorbital lacrimal gland, which in turn, inhibits the release of acetylcholine into the nerve junction, thereby blunting the secretion of tears. Indeed, our data confirm the presence of robust ocular surface damage in the setting of reduced tear production. More important was the finding that tissue levels of IGFBP-3 were similarly reduced in the mouse corneal epithelium. While we have previously shown that IGFBP-3 is present in human tears and is altered in disease, given the small surface area and minute tear volume, the analysis of IGFBP-3 in mouse tear fluid in this study was not feasible [
39,
40,
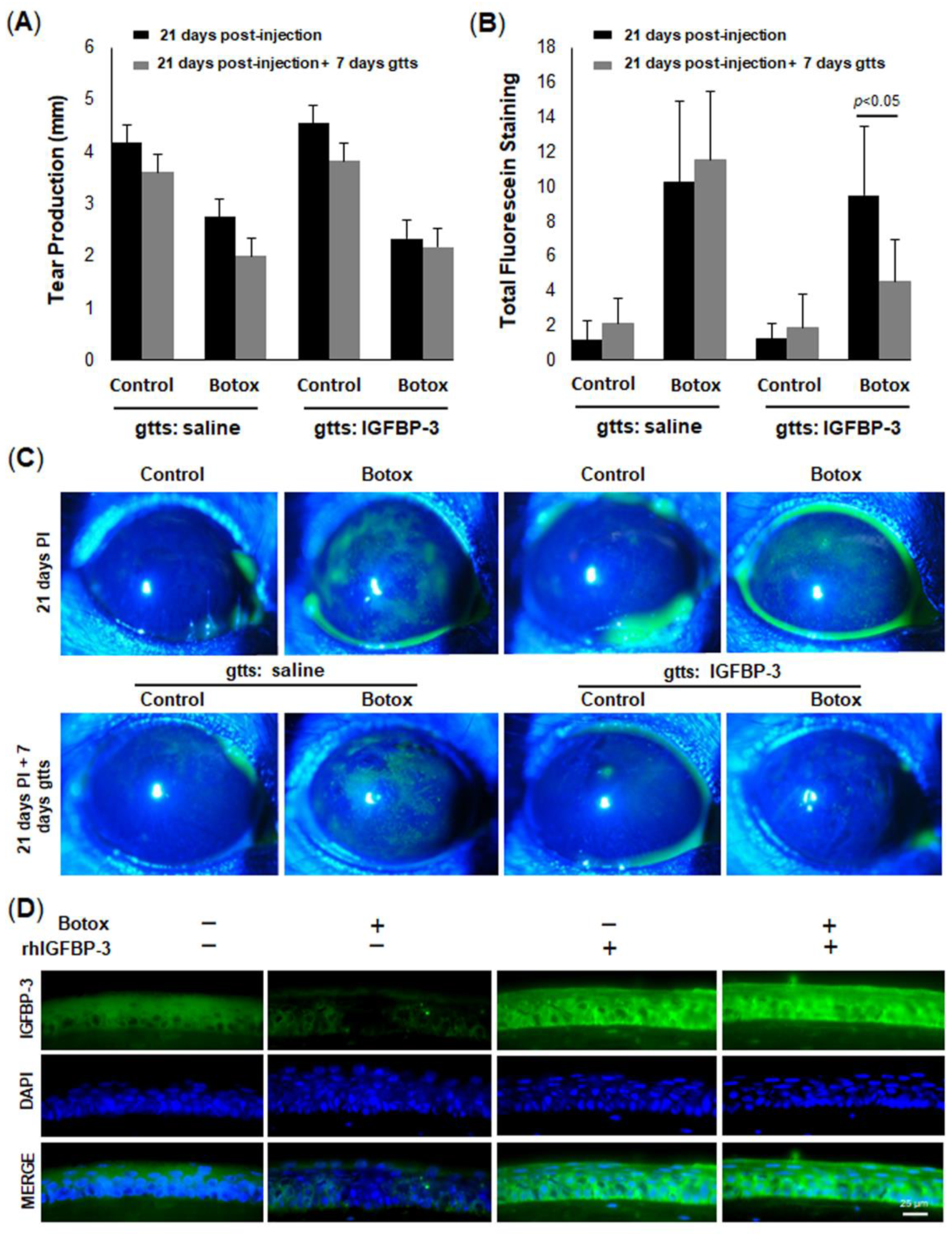
55]. We did show, however, that the topical administration of IGFBP-3 to the mouse cornea was cytoprotective and significantly decreased corneal surface damage, despite the reduction in tear secretion. Further studies are now needed to evaluate the in vivo effects of IGFBP-3 on mitochondria and metabolic adaptation. In addition, it is unknown whether the protective effects on the mouse cornea are transient in nature or whether prolonged hyperfusion will lead to an accumulation of mitochondrial defects due to impaired mitophagy.
In summary, these findings establish a new functional role for IGFBP-3 in the promotion of mitochondrial hyperfusion and metabolic activity in cells exposed to hyperosmolar stress. These data also support the idea that IGFBP-3 modulates a biphasic response during stress. This includes an early, immediate response by trafficking IGF-1R to the mitochondria to stabilize the organelle, while the nuclear translocation of IGFBP-3 induces metabolic adaptation through increased protein translation, mitochondrial fusion, and oxidative phosphorylation. Future studies will focus on the mechanistic determination of how IGFBP-3 exerts these adaptive effects, whether mitochondrial hyperfusion occurs transiently during metabolic adaptation, or whether it is sustained with potential long-term cellular effects. While current therapeutic interventions for DED are focused on lowering inflammation, these studies could lead to an entirely new class of therapeutics for patients with severe DED and other non-ocular tissues with underlying mitochondrial and metabolic dysfunction.
4. Materials and Methods
4.1. Cell Lines and Culture
Two telomerase immortalized human epithelial cell lines were used in these studies. This includes the human telomerized corneal epithelial (hTCEpi) cell line established and characterized by our laboratory and human conjunctival (HCjE) cells (immortalized with hTERT, mutant Cdk4, and dominant negative p53), which were provided by Drs. Pablo Argüeso (Schepens Eye Research Institute, Harvard Medical School, Boston, MA, USA) and Ajay Sharma (School of Pharmacy, Chapman University, Orange, CA, USA) [
56,
57,
58]. Both cell lines were cultured in serum-free keratinocyte basal media (KBM) with growth factor supplements (KGM, Keratinocyte Growth Medium 2, PromoCell, VWR, Radnor, PA, USA). Due to low levels of calcium in KBM, cultures were further supplemented with a calcium chloride solution (PromoCell, VWR, Radnor, PA, USA) to a final concentration of 0.15 mM. Cultures were maintained at 37 °C with 5% CO
2. Primary cultures were established from human donor corneas obtained from Tissue Transplant Services at UT Southwestern Medical Center. To accomplish this, human corneal epithelial cells (HCECs) were harvested as previously described [
48]. CnT20 cell culture media enriched for progenitor cell culture (Zen Bio, Research Triangle Park, NC, USA) were used for initial cultures. After the first passage, HCECs were transitioned to serum-free KGM media, described above. The osmolarity of the isotonic media was approximately 330 mOsm, as specified by manufacturer data. For hyperosmolarity experiments, hyperosmolar medium was made by the addition of NaCl (Thermo Fisher, St. Louis, MO, USA) to reach the desired osmolarity. Recombinant human (rh)IGFBP-3 (500 ng/mL) was used in select experiments to determine the effects of exogenous IGFBP-3 on all cell types.
4.2. Reagents
rhIGFBP-3 was acquired from Sino Biological (Chesterbrook, PA, USA). The lyophilized protein was re-suspended using ultrapure water. Antibodies used for immunoblotting and immunofluorescence included: a rabbit polyclonal anti-IGF-1Rβ #3027, a rabbit monoclonal anti-mitofusion-2 #9482, a rabbit monoclonal anti-mitofusion-1 #14739, a rabbit monoclonal anti-OPA1 #67589, a rabbit monoclonal anti-COX IV #4850, a rabbit polyclonal anti-VDAC1 #4661 (Cell Signaling, Danvers, MA, USA); a mouse monoclonal anti-GAPDH #sc-66163, a mouse monoclonal anti-β-actin #sc-47778 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); and a mouse monoclonal anti-puromycin #MABE343 (EMB Millipore, Burlington, MA, USA). Secondary antibodies used for immunofluorescence were anti-rabbit IgG conjugated to Alexa Fluor 488 and anti-mouse IgG conjugated to Alexa Fluor 555 (Cell Signaling, Danvers, MA, USA). For immunoblotting, secondary antibodies were goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP) #170-6515 and goat anti-mouse conjugated to HRP #170-6516 (Bio-rad, Hercules, CA, USA).
4.3. Determination of Cell Number
hTCEpi cells were seeded in 24-well tissue culture plates in KGM at 70% confluence and allowed to adhere overnight. Media were then removed and replaced with isotonic KBM or KBM supplemented with NaCl to a final concentration of 450 mOsm with or without rhIGFBP-3 and cultured for an additional 6 or 24 h. Cell counts and images were captured using a Celigo imaging cytometer (Nexcelom, Lawrence, MA, USA). Cell count experiments were performed in quadruplicate and repeated a minimum of two additional times.
4.4. Cell Cycle Assay
Cell proliferation was measured by seeding hTCEpi cells in a 24-well plate in KGM. Following this, media were replaced with isotonic KBM or KBM supplemented with NaCl to 450 mOsm with or without 500 ng/mL rhIGFBP-3. Cells were incubated in these treatments for 6 or 24 h, as indicated. At the appropriate time points, cells were washed with phosphate-buffered saline and fixed for 15 min with ice-cold ethanol. After fixation, cells were stained with propidium iodide (Thermo Fisher, St. Louis, MO, USA) and imaged using a Celigo imaging cytometer (Nexcelom, Lawrence, MA, USA). Each experiment was repeated a minimum of two additional times.
4.5. Enzyme-Linked Immunoassay (ELISA)
Cells were plated using KGM in six-well culture dishes and allowed to adhere overnight. Media were removed and cells were treated with KBM or KBM 450 mOsm media with or without rhIGFBP-3 for 24 h. Media were collected and concentrated using protein concentrators containing a polyethersulfone membrane (3K MWCO; Millipore, Burlington, MA, USA). Cell lysates were harvested using buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and a protease and phosphatase inhibitor cocktail (Thermo Fisher, Rockford, IL, USA). Whole-cell lysates were kept on ice and vortexed intermittently for thirty minutes. Protein concentration was measured using a Qubit 3.0 Fluorometer (Thermo Fisher, Waltham, MA, USA). A human IGFBP-3 Quantikine enzyme-linked immunoassay (ELISA, R&D systems, Minneapolis, MN, USA) was used to analyze IGFBP-3 levels in conditioned media and whole-cell lysates. Measurements were obtained using a BioTek Synergy 2 Multi-Mode Microplate Reader (Thermo Fisher, Waltham, MA, USA) All samples were run in triplicate and repeated a minimum of two additional times.
4.6. Real-Time Metabolic Studies
A Seahorse Metabolic Analyzer XFp (Agilent Technologies, Santa Clara, CA, USA) was used to acquire real-time measurements of cellular oxygen consumption rate (OCR) and intracellular acidification rate (ECAR). hTCEpi cells were seeded into Seahorse XFp miniplates and incubated overnight at 37 °C, 5% CO2. The media were then removed, and cells were treated with KBM (basal) or KBM with increasing osmolarity levels (375, 400, or 450 mOsm) for another 24 h at 37 °C, 5% CO2. After 24 h, the media were removed and Seahorse XF base medium containing 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose (pH 7.4) was added. Plates were then incubated at 37 °C in a non-CO2 incubator for 1 h. Mitochondrial metabolism was analyzed using a Seahorse XFp Cell Mito Stress Test Kit (Agilent Technologies, Santa Clara, CA, USA). During the test, measurements were obtained every 6 min for a total of 94 min. At 20 min, 10 μM oligomycin was added to inhibit ATP synthase. This was followed by an injection of 10 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) at 50 min, in order to uncouple the proton gradient and allow for maximal respiration. Lastly, an injection of rotenone and antimycin A was added at 80 min, which inhibited complex 1 and 3 of the electron transport chain, thereby disabling mitochondrial respiration, in order to quantify non-mitochondrial respiration. All data were analyzed using the manufacturers’ Wave software, version 2.3.0. The following equations were utilized: spare respiratory capacity = (maximal respiration)/(basal respiration) × 100; coupling efficiency = (ATP-linked respiration rate)/(basal respiration rate) × 100. All six wells were used as technical replicates for each tech condition. The entire experiment was repeated a minimum of two additional times. Immediately after completing the assay, all wells were normalized for cell number using a Celigo Image Cytometer (Nexcelom Bioscience, Lawrence, MA, USA). An Amplex Red Enzyme Assay (Thermo Fisher, Rockford, I) was further used to measure H2O2 production. This is due to the production of fluorescent resorufin when it reacts with H2O2. Briefly, hTCEpi cells were seeded onto a 96-black-well plate and allowed to adhere overnight. Media were then changed, and cells were treated for 24 h in isotonic KBM or KBM with increased osmolarity (450 mOsm) with or without rhIGFBP-3. Fluorescence was measured at excitation/emission maxima of 530/590 nm on a Cellometer K2 Fluorescent Viability Cell Counter (Nexcelom, Lawrence, MA, USA). All assays were conducted in triplicate and repeated a minimum of two additional times.
4.7. Transmission Electron Microscopy (TEM)
hTCEpi cells were seeded onto 35 mm glass-bottom dishes (MatTek Corporation, Ashland, MA, USA) containing KGM and allowed to adhere overnight. Cells were treated for 24 h in KBM with or without rhIGFBP-3. Following treatment, 2.5% glutaraldehyde/0.1 M cacodylate buffer pH 7.4 was added to the cells for 15 min at room temperature to allow for fixation. Next, cells were washed three times for five minutes with 0.1 M sodium cacodylate buffer. Following washing, cells were post-fixed in 1% osmium tetroxide and 0.8% K
3[Fe(CN
6)] in 0.1 M sodium cacodylate buffer for 1 h at room temperature and then rinsed with water. Cells were next stained overnight en bloc with 2% aqueous uranyl acetate and then washed with water. Cells were then dehydrated through exposure to increasing concentrations of ethanol and infiltrated with embed-812 resin and polymerized at 60 °C overnight. Once embedded, cell blocks were sectioned on a Leica Ultracut UCT [
7] ultramicrotome (Leica Microsystems, Heidelberg, Germany) using a diamond knife (Diatome, Hatfield, PA, USA) and positioned on copper grids. Finally, samples on copper grids were post-stained with 2% uranyl acetate in water and lead citrate. Imaging was performed using a JEOL 1400 Plus (JEOL) equipped with a LaB
6 source using a voltage of 120 kV.
4.8. Polymerase Chain Reaction
Cells were plated using KGM in six-well culture dishes and allowed to adhere overnight. Media were removed, and cells were treated with KBM or KBM 450 mOsm media with or without rhIGFBP-3 for 24 h. Following treatment, a RNeasy kit (Qiagen, Germantown, MD, USA) was used to extract RNA according to the manufacturer protocol. A gDNA Wipeout Buffer eliminated residual genomic DNA (Qiagen, Germantown, MD, USA). A Nanodrop OneC (Thermo Fisher, Rockford, IL, USA) was used to quantify the level of RNA. A QuantiTect Reverse Transcription Kit (Qiagen, Germantown, MD, USA) was used to reverse transcribe 1 μg mRNA into cDNA. A QuantiFast SYBR Green PCR kit (Qiagen, Germantown, MD, USA) was used to perform 40 cycles of real-time PCR on a QuantStudio 6 Flex Real Time PCR machine (Applied Biosystems, Foster City, CA, USA). For real-time PCR, 100 ng cDNA was amplified by using 1 µM of each QuantiTect Primer Assay (Qiagen, Germantown, MD, USA) for Hs_EIF2AK1 (QT01018920), Hs_PTGS2 (QT00040586) and Hs_LOC100128596 (QT02432626). These correspond to a mitochondrial encoded gene, Cyclooxygenase 2 (COX2), which was analyzed along with two nuclear encoded genes, Eukaryotic Initiation Factor 2 (EIF2) and NADH Dehydrogenase. Levels of COX2 were normalized to each of the two nuclear encoded genes for normalization. Total reaction volume was 10 µL. Samples were plated in triplicate for all experiments. Water was used as a no-template control. All controls were performed in parallel. Experiments were repeated a minimum of two additional times. Data were analyzed using the 2-ΔΔCT method.
4.9. siRNA Knockdown of IGF-1R
hTCEpi cells were seeded at 50–60% confluence into six-well plates and allowed to adhere overnight. Cells were then transfected with double-stranded inhibitory RNA oligonucleotides to IGF-1R or IGFBP-3 (FlexiTube GeneSolution, IGF-1R #GS3480, Qiagen, Germantown, MD, USA), using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) in antibiotic-free KGM. An amount of 12 pmol of siRNA oligonucleotides targeting IGF-1R or non-targeting controls were added to 100 µL KGM and incubated at room temperature for five minutes. The siRNA was combined with 2 µL Lipofectamine diluted in 100 µL KGM. This mixture was incubated for fifteen minutes at room temperature. Next, the transfection mixture was added to hTCEpi cells containing 1 mL of KGM media. Samples were then incubated for 24 h prior to treatment. Transfection media were removed, and cells were cultured in 450 mOsm KBM for another 24 h, with IGFBP-3 confirming the specificity of the antibody for IGF-1R in the mitochondrial fraction. Allstars negative control siRNA was used as the non-targeting control (Qiagen, Germantown, MD, USA).
4.10. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis and Immunoblotting
For immunoblotting of whole-cell lysates, a lysis buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and a protease and phosphatase inhibitor cocktail (Thermo Fisher, Rockford, IL, USA) was used to directly lyse adherent epithelial cells in 6-well culture plates. Samples were kept on ice and vortexed intermittently for 30 min, followed by centrifugation for 5 min at 12,000 rpm at 4 °C (BioRad, Hercules, CA, USA). After centrifugation, the supernatants were collected and protein concentration was quantified using a Qubit 3.0 Fluorometer (Thermo Fisher, Rockford, IL, USA). A 2× sample buffer pH 6.8 containing 65.8 mM Tris-HCl, 26.3% (w/v) glycerol, 2.1% SDS, 5.0% β-mercaptoethanol and 0.01% bromophenol blue was added to the sample, which was then boiled for five minutes (Bio-rad, Hercules, CA, USA). Next, boiled samples were electrophoresed through a 4–15% precast linear gradient polyacrylamide gel (Bio-rad, Hercules, CA, USA). Once electrophoresis was complete, gels were transferred onto a polyvinyl difluoride (PVDF) membrane (Millipore, Temecula, CA, USA). Membranes were then blocked with 5% non-fat milk for one hour at room temperature (Bio-rad, Hercules, CA, USA). After blocking, membranes were washed three times for five minutes with PBS. Membranes were incubated overnight in primary antibody at 4 °C and then washed again three times for five minutes. Membranes were incubated with secondary antibody at room temperature for one hour. Secondary antibody was either anti-mouse or anti-rabbit secondary antibody (Santa Cruz, CA, USA). An Amersham Imager 600 (Amersham Biosciences, Piscataway, NJ, USA) was used to image the protein present on the membrane with ECL Plus Prime Detection Reagent (Amersham Biosciences, Piscataway, NJ, USA). β-actin or GAPDH were used as loading controls for normalization. ImageQuant TL Toolbox v8.1 software was used to analyze and quantify proteins (Amersham Bioscience, Piscataway, NJ, USA). To measure global protein translation, a SUnSET Assay was used. hTCEpi cells were seeded into a six-well plate at 70% confluence and allowed to adhere overnight. The media were then replaced with isotonic KBM (basal) or 450 mOsm KBM with or without rhIGFBP-3. Two sets of each group were treated, and cells were cultured for 22 h under these conditions. At 22 h, one set from each group was treated with cycloheximide solution (Sigma, St. Louis, MO, USA). All groups were then immediately treated with puromycin dihydrochloride (Sigma, St. Louis, MO, USA). Cells were cultured for an additional two hours. Cells were then lysed and blotted as described above.
4.11. Nuclear and Mitochondrial Fractionation with Immunoblotting
For experiments quantifying the intracellular localization of IGFBP-3 and IGF-1R, hTCEpi cells were subjected to subcellular fractionation. Trypsin-ethylenediaminetetraacetic acid (EDTA, Gibco, St. Louis, MO, USA) was used to dissociate adherent cells after 2 or 24 h of treatment in isotonic KBM or 450mOsm KBM with or without IGFBP-3. Cells were then centrifuged and washed with PBS prior to use. To isolate the mitochondrial fraction, a mitochondrial fractionation kit (Thermo Fisher, Rockford, IL, USA) was used. A nuclear fractionation kit (Thermo Fisher, Rockford, IL, USA) was used to separate the cytoplasmic, insoluble and soluble nuclear fractions. Fractions were boiled with 2× sample buffer and electrophoresed through a 4–15% polyacrylamide gel (BioRad, Hercules, CA, USA). Immunoblotting was performed as described above. A voltage-dependent anion channel (VDAC) was used as a mitochondrial control. Histone H3 was used as a control for the insoluble nuclear fraction and SP1 for the soluble nuclear fractionation.
4.12. Botulinum-Toxin-B-Induced Dry Eye Disease Mouse Model
A total of 104 mice were used in this study. All mice were C57BL6 Type N strain aged 6 to 8 weeks (Charles River, Wilmington, MA, USA). An aqueous-deficient dry eye model was used according to a previously reported method [
59,
60]. All mice for these studies were housed at a relatively stable temperature (21–24 °C) and humidity. A sterile 33G needle was used to inject the lacrimal gland with botulinum toxin-B (Botox, 20 mU in 0.05 mL 0.9% saline) or the vehicle control (0.05 mL of 0.9% saline). Only the right eye was used for these experiments due to the potential for sympathetic responses between eyes. Mice were randomly assigned to the Botox or control group and there were an equal number of male and females in each group. All mice were screened prior to injection for ocular surface abnormalities. To determine the effect of topical treatment of rhIGFBP-3 in DED, 10 μL of a solution containing 500 ng/mL of rhIGFBP-3 was applied on the surface of the eye. Either rhIGFBP-3 or the vehicle control (sterile saline) were applied twice a day on alternating days for the final seven days prior to tissue collection and analysis.
4.13. Clinical Measurements
To quantify ocular surface damage, fluorescein staining was performed at Day 0, 7, 14, and 28 to determine the severity of DED. Fluorescein sodium ophthalmic strips (1 mg, BioGlo, Scottsdale, AZ, USA) were used for these experiments. Examination of the ocular surface was performed using a slit-lamp biomicroscope with a cobalt blue light and yellow Wratten filter. Photographs of each mouse were obtained using a Sony Cyber Shot camera (New York, NY, USA) fitted to the slit-lamp eye piece. For fluorescein grading, the eye was subdivided into five quadrants that were scored separately by a blinded observer. Each quadrant was scored using a scale of 0–3 with 0 indicating no staining and 3 indicating severe ocular surface damage [
61]. The total score was the sum of the score from all five quadrants. A phenol red thread test was used to determine aqueous tear production (Zone-Quick, Oasis, CA, USA). The tip of the thread was placed in the lateral canthus of the right eye for 15 s. The length of thread that had turned red was measured using a micron ruler.
4.14. Immunofluorescence
Mouse corneas and hTCEpi cells were used for immunofluorescent assays. For the in vitro studies, hTCEpi cells were seeded onto 35 mm glass-bottom dishes (MatTek Corporation, Ashland, MA, USA). siRNA was used to knockdown IGFBP-3 as described and cells were incubated for 24 h. Cells were then cultured in KBM (basal) with or without rhIGFBP-3 for an additional 24 h. After, cells were washed twice with cold PBS and fixed in 1% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA, USA) in PBS for 10 min. Following this, cells were washed with PBS and then permeabilized in 0.1% Triton X-100 in PBS for 10 min. Samples were then washed three times for five minutes with PBS. Next, cells were blocked using 0.5% bovine serum albumin (Sigma, St. Louis, MO, USA) in PBS for 30 min. Cells were incubated overnight at 4 °C using primary antibodies directed against MFN1, MFN2, or IGFBP-3 diluted in 0.1% bovine serum albumin (Sigma, St. Louis, MO, USA). After, each sample was washed in PBS and stained with an anti-rabbit IgG conjugated to Alexa Fluor 488 for 1 h at room temperature. Prolong gold anti-fade reagent containing DAPI was used for mounting and nuclear staining (Invitrogen, Carlsbad, CA, USA). In order to analyze IGFBP-3 expression in the mouse corneal epithelium, we performed both ELISA (detailed earlier) and immunofluorescence. For the latter, whole-mouse globes were excised, fixed in 1% paraformaldehyde, and embedded in Tissue-Plus™ O.C.T. Compound Tissue-Plus™ embedding medium (Thermo Fischer, Waltham, MA, USA) and frozen in a −80 °C freezer. Cryostat-sectioned tissues were washed three times with PBS for five minutes. Following this, tissue sections were rewashed with PBS and permeabilized using ice-cold acetone. After washing, tissue sections were blocked with 0.5% BSA in PBS and then stained using primary and secondary antibodies as described above. Cells were imaged on a Leica SP8 laser scanning confocal microscope (Leica Microsystems, Heidelberg, Germany) using a 63× oil objective. In order to prevent spectral crosstalk between channels, images were sequentially scanned.
4.15. Statistics
All data are presented as mean ± standard deviations. For the comparison between two groups, a Student’s t-test was utilized. For comparisons between more than two groups, a one-way ANOVA was used. For ANOVA analysis, an appropriate post hoc multiple comparison test was performed. Statistical significance was set at p < 0.05.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








