
shRNAs Targeting a Common KCNQ1 Variant Could Alleviate Long-QT1 Disease Severity by Inhibiting a Mutant Allele


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
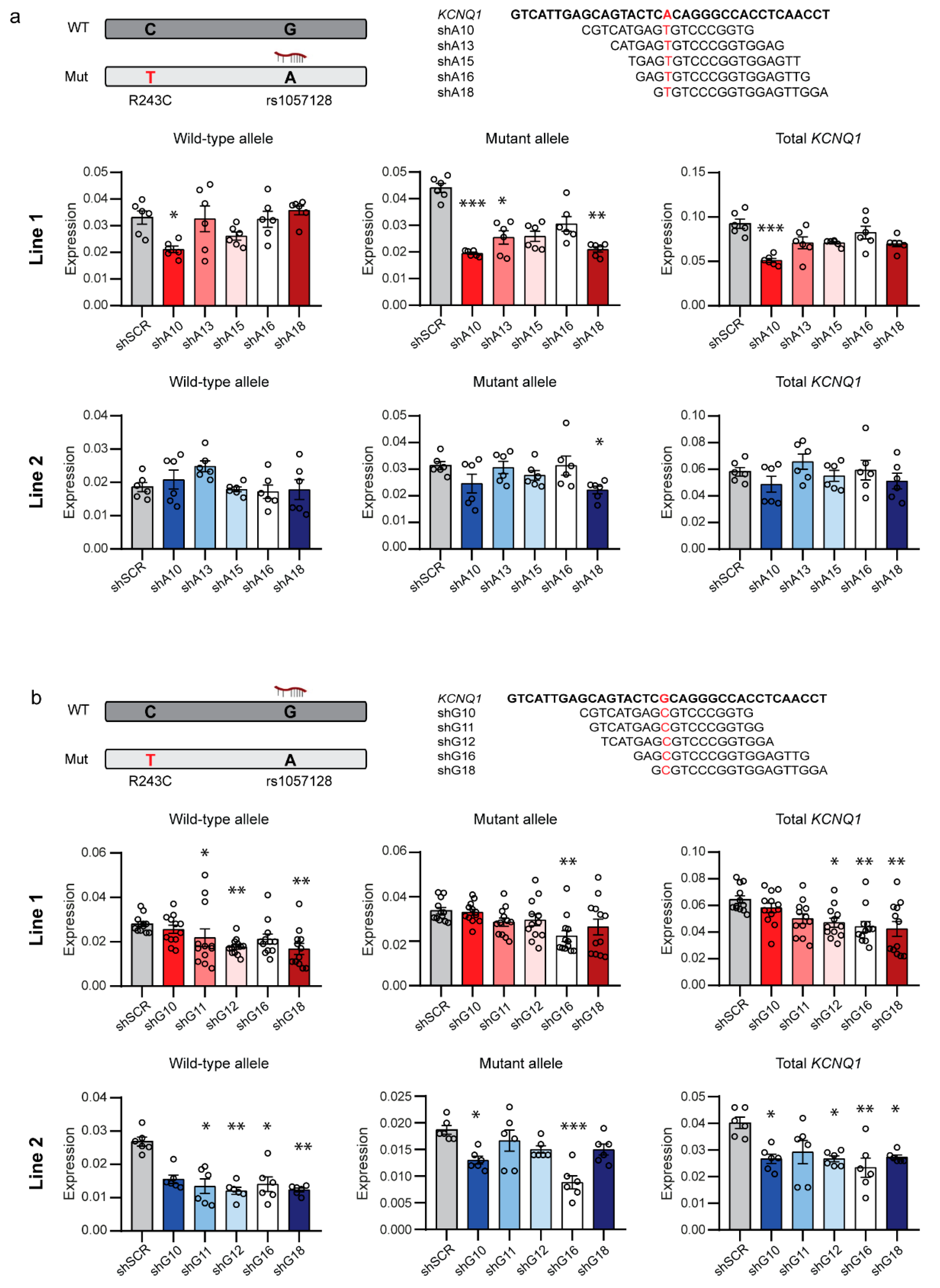
2.1. Allele-Specific Targeting of Common SNPs in KCNQ1
2.2. Allele-Specific Targeting of SNPs in the 3′UTR of KCNQ1
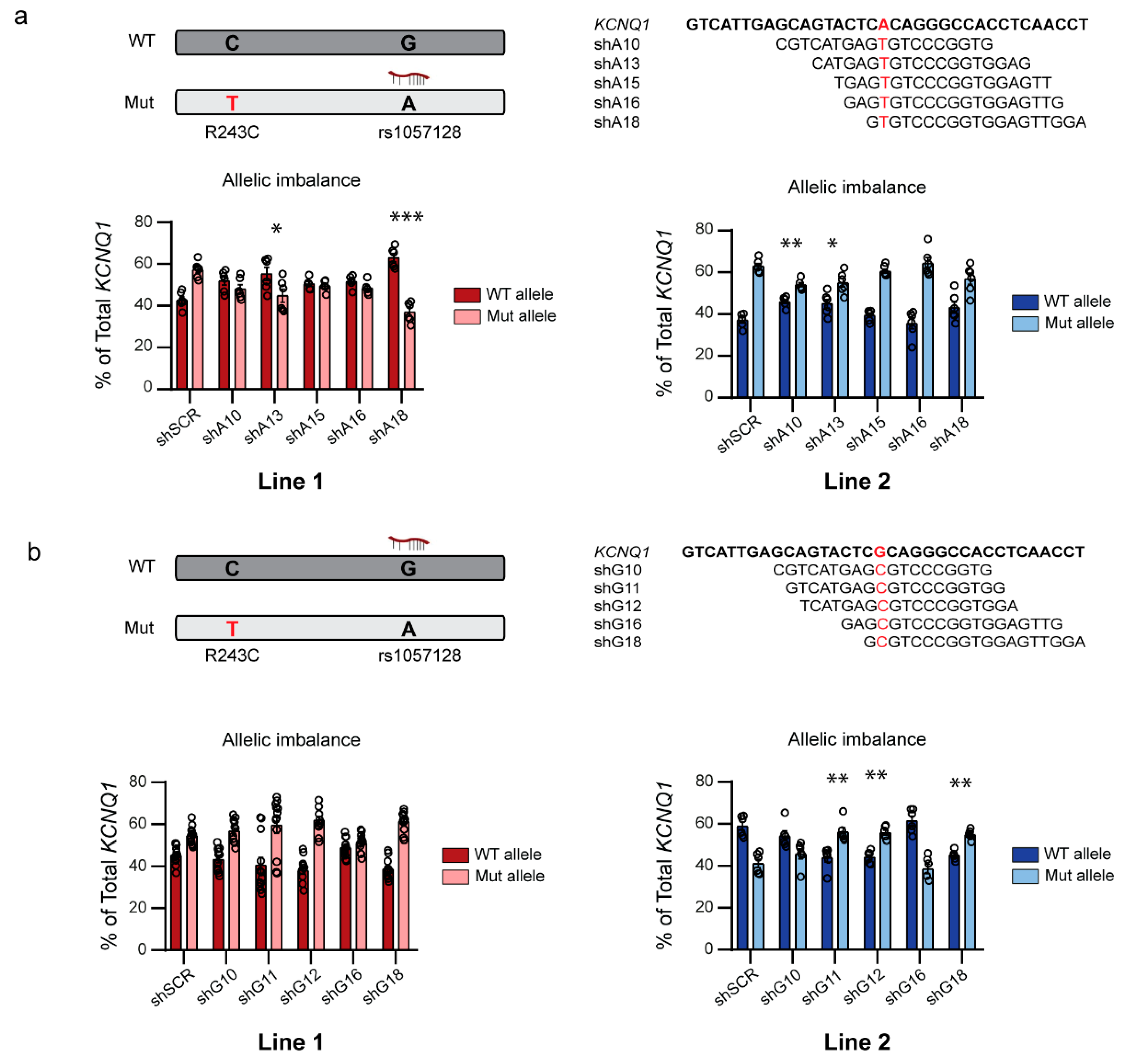
2.3. Allele-Specific shRNAs Affect the Allelic Balance in hiPSC-CMs
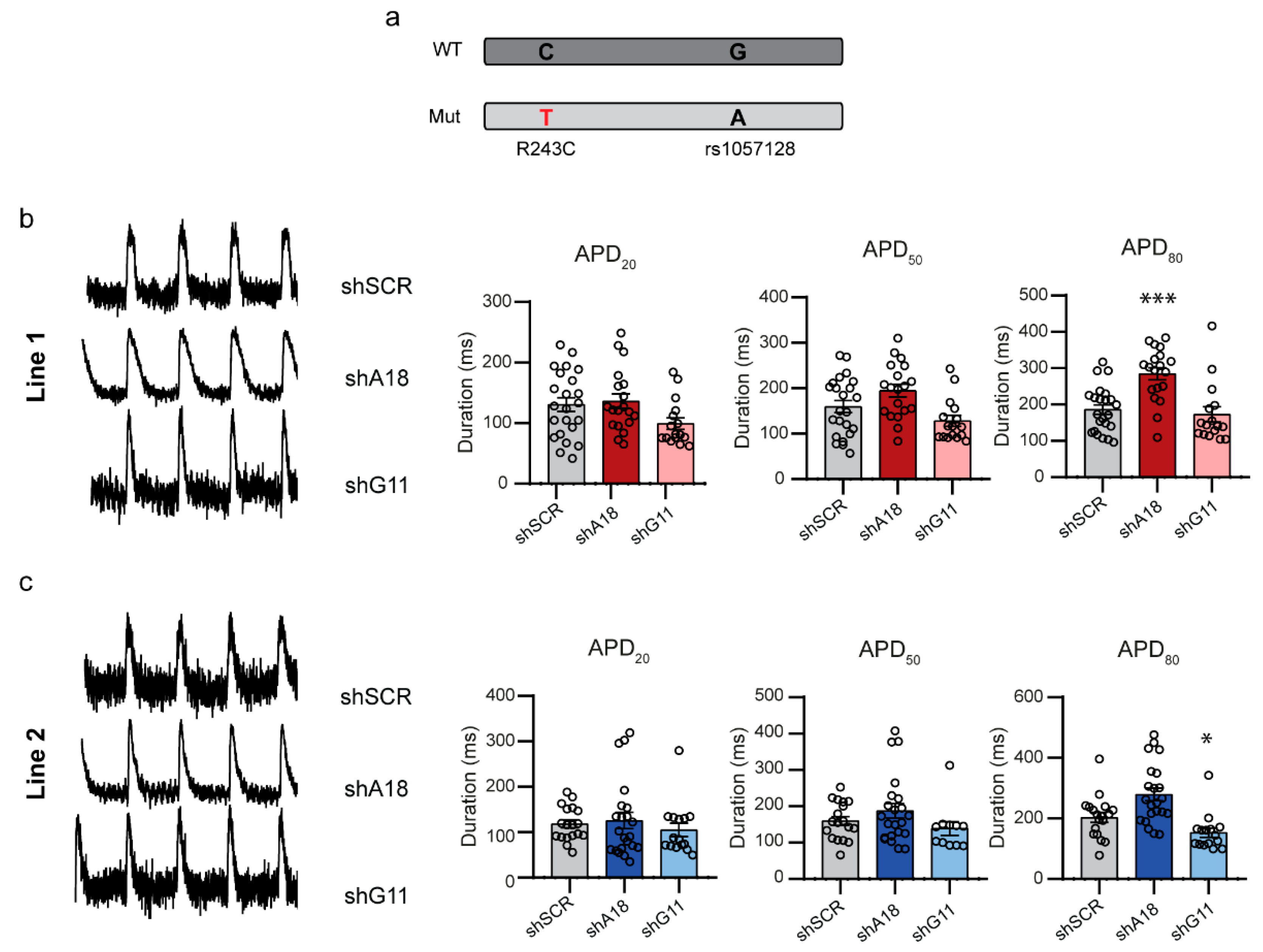
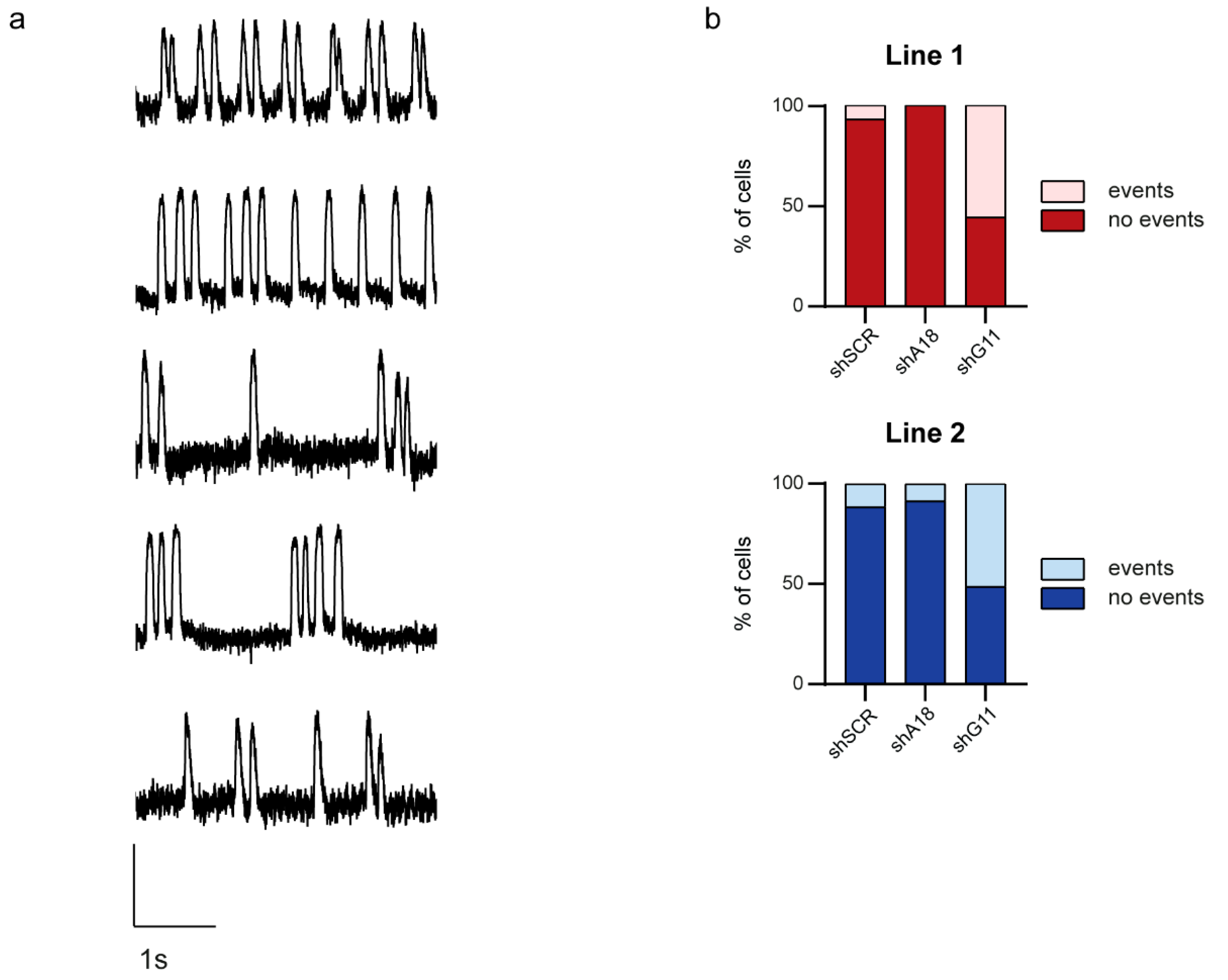
2.4. Specific Downregulation of the Mutant KCNQ1 Allele Prevents the Occurrence of Arrhythmic Events
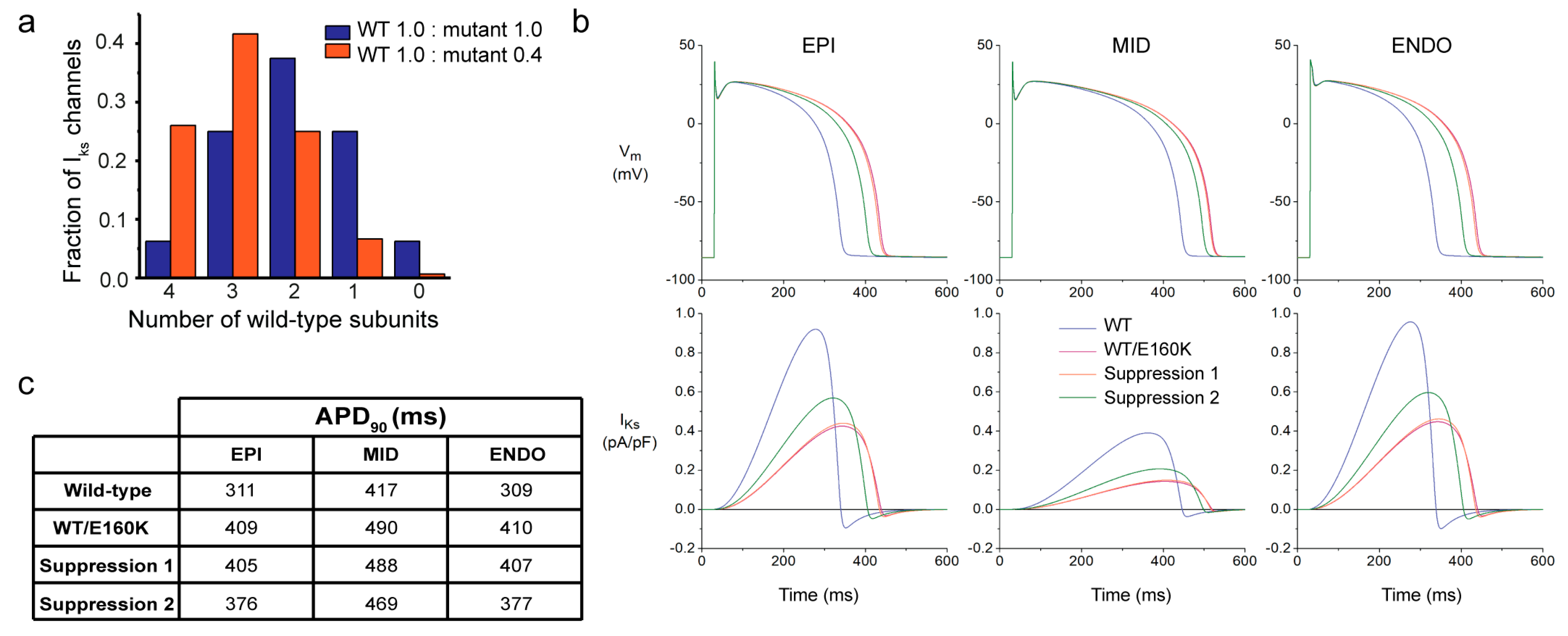
2.5. Computer Simulations Demonstrate the Applicability of Allele-Specific Inhibition in an Adult Human Cardiomyocyte Model
3. Discussion
4. Materials and Methods
4.1. Human iPSC Generation
4.2. Human iPSC Culture
4.3. Karyotype Analysis
4.4. Cardiac Differentiation of hiPSC
4.5. In Vitro Trilineage Differentiation Potential
4.6. Immunocytochemistry
4.7. Plasmid Generation
4.8. Virus Production
4.9. hiPSC-CMs Infection
4.10. RNA Isolation
4.11. qRT-PCR
4.12. ArcLight Measurements
4.13. Computer Simulations
4.14. Statistics
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Zareba, W.; Moss, A.J. Long QT Syndrome. Curr. Probl. Cardiol. 2008, 33, 629–694. [Google Scholar] [CrossRef]
- Mizusawa, Y.; Horie, M.; Wilde, A.A. Genetic and clinical advances in congenital long QT syndrome. Circ. J. 2014, 78, 2827–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Horr, S.; Moss, A.J.; Lopes, C.M.; Barsheshet, A.; McNitt, S.; Zareba, W.; Andrews, M.L.; Robinson, J.L.; Locati, E.H.; et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J. Am. Coll. Cardiol. 2011, 57, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [Green Version]
- Moss, A.J.; Zareba, W.; Hall, W.J.; Schwartz, P.J.; Crampton, R.S.; Benhorin, J.; Vincent, G.M.; Locati, E.H.; Priori, S.G.; Napolitano, C.; et al. Effectiveness and limitations of β-blocker therapy in congenital long-QT syndrome. Circulation 2000, 101, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Waddell-Smith, K.E.; Earle, N.; Skinner, J.R. Must every child with long QT syndrome take a beta blocker? Arch. Dis. Child 2015, 100, 279–282. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, C.E.; Waddell-Smith, K.E.; Skinner, J.R.; Broadbent, E. Predictors of β-blocker adherence in cardiac inherited disease. Open Heart 2018, 5, e000877. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013, 15, 1389–1406. [Google Scholar] [CrossRef]
- Shalaby, F.Y.; Levesque, P.C.; Yang, W.P.; Little, W.A.; Conder, M.L.; Jenkins-West, T.; Blanar, M.A. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation 1997, 96, 1733–1736. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kuenze, G.; Smith, J.A.; Taylor, K.C.; Duran, A.M.; Hadziselimovic, A.; Meiler, J.; Vanoye, C.G.; George, A.L., Jr.; Sanders, C.R. Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci. Adv. 2018, 4, eaar2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.; Wimmer, A.B.; Karle, C.A.; Licka, M.; Alter, M.; Khalil, M.; Ulmer, H.E.; Kathöfer, S.; Kiehn, J.; Katus, H.A.; et al. Dominant-negative IKs suppression by KCNQ1-ΔF339 potassium channels linked to Romano-Ward syndrome. Cardiovasc. Res. 2005, 67, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Amin, A.S.; Giudicessi, J.R.; Tijsen, A.J.; Spanjaart, A.M.; Reckman, Y.J.; Klemens, C.A.; Tanck, M.W.; Kapplinger, J.D.; Hofman, N.; Sinner, M.F.; et al. Variants in the 3’ untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur. Heart J. 2012, 33, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Dainis, A.; Zaleta-Rivera, K.; Ribeiro, A.; Chang, A.C.H.; Shang, C.; Lan, F.; Burridge, P.W.; Liu, W.R.; Wu, J.C.; Chang, A.C.Y.; et al. Silencing of MYH7 ameliorates disease phenotypes in human iPSC-cardiomyocytes. Physiol. Genom. 2020, 52, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Zaleta-Rivera, K.; Dainis, A.; Ribeiro, A.J.S.; Cordero, P.; Rubio, G.; Shang, C.; Liu, J.; Finsterbach, T.; Parikh, V.N.; Sutton, S.; et al. Allele-specific silencing ameliorates restrictive cardiomyopathy attributable to a human myosin regulatory light chain mutation. Circulation 2019, 140, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Bongianino, R.; Denegri, M.; Mazzanti, A.; Lodola, F.; Vollero, A.; Boncompagni, S.; Fasciano, S.; Rizzo, G.; Mangione, D.; Barbaro, S.; et al. Allele-specific silencing of mutant mRNA rescues ultrastructural and arrhythmic phenotype in mice carriers of the R4496C mutation in the ryanodine receptor gene (RYR2). Circ. Res. 2017, 121, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yang, X.; Huang, X.; Huang, C.; Sun, H.H.; Jin, L.; Xu, W.; Mao, H.; Guo, J.; Zhou, J.; et al. RNA interference targeting E637K mutation rescues hERG channel currents and restores its kinetic properties. Heart Rhythm 2013, 10, 128–136. [Google Scholar] [CrossRef]
- Matsa, E.; Dixon, J.E.; Medway, C.; Georgiou, O.; Patel, M.J.; Morgan, K.; Kemp, P.J.; Staniforth, A.; Mellor, I.; Denning, C. Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur. Heart J. 2014, 35, 1078–1087. [Google Scholar] [CrossRef] [Green Version]
- Dotzler, S.M.; Kim, C.S.J.; Gendron, W.A.C.; Zhou, W.; Ye, D.; Bos, J.M.; Tester, D.J.; Barry, M.A.; Ackerman, M.J. Suppression-replacement KCNQ1 gene therapy for type 1 long QT syndrome. Circulation 2021, 143, 1411–1425. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Qiao, R.; Zhao, D.; Zhang, T.; Li, Y.; Yi, F.; Lai, F.; Hong, J.; Ding, X.; Yang, Z.; et al. Profiling of mismatch discrimination in RNAi enabled rational design of allele-specific siRNAs. Nucleic Acids Res. 2009, 37, 7560–7569. [Google Scholar] [CrossRef] [Green Version]
- Franqueza, L.; Lin, M.; Shen, J.; Splawski, I.; Keating, M.T.; Sanguinetti, M.C. Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 1999, 274, 21063–21070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matavel, A.; Medei, E.; Lopes, C.M. PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels 2010, 4, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsheshet, A.; Goldenberg, I.; J, O.U.; Moss, A.J.; Jons, C.; Shimizu, W.; Wilde, A.A.; McNitt, S.; Peterson, D.R.; Zareba, W.; et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: Implications for mutation-specific response to β-blocker therapy in type 1 long-QT syndrome. Circulation 2012, 125, 1988–1996. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Tokunaga, K.; Hohjoh, H. Influence of assembly of siRNA elements into RNA-induced silencing complex by fork-siRNA duplex carrying nucleotide mismatches at the 3’- or 5’-end of the sense-stranded siRNA element. Biochem. Biophys. Res. Commun. 2005, 329, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Han, Z.; Platisa, J.; Wooltorton, J.R.; Cohen, L.B.; Pieribone, V.A. Single action potentials and subthreshold electrical events imaged in neurons with a fluorescent protein voltage probe. Neuron 2012, 75, 779–785. [Google Scholar] [CrossRef] [Green Version]
- Mazzanti, A.; Maragna, R.; Vacanti, G.; Monteforte, N.; Bloise, R.; Marino, M.; Braghieri, L.; Gambelli, P.; Memmi, M.; Pagan, E.; et al. Interplay between genetic substrate, QTc duration, and arrhythmia risk in patients with long QT syndrome. J. Am. Coll. Cardiol. 2018, 71, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Vanoye, C.G.; Desai, R.R.; Fabre, K.L.; Gallagher, S.L.; Potet, F.; DeKeyser, J.M.; Macaya, D.; Meiler, J.; Sanders, C.R.; George, A.L., Jr. High-throughput functional evaluation of KCNQ1 decrypts variants of unknown significance. Circ. Genom. Precis. Med 2018, 11, e002345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, L.; Priori, S.G.; Napolitano, C.; Surewicz, K.A.; Dennis, A.T.; Memmi, M.; Schwartz, P.J.; Brown, A.M. Mechanisms of IKs suppression in LQT1 mutants. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H3003–H3011. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Sakaguchi, T.; Takenaka, K.; Toyoda, F.; Tsuji, K.; Matsuura, H.; Horie, M. A trafficking-deficient KCNQ1 mutation, T587M, causes a severe phenotype of long QT syndrome by interfering with intracellular hERG transport. J. Cardiol. 2019, 73, 343–350. [Google Scholar] [CrossRef]
- Jiang, J.; Wakimoto, H.; Seidman, J.G.; Seidman, C.E. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 2013, 342, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trochet, D.; Prudhon, B.; Beuvin, M.; Peccate, C.; Lorain, S.; Julien, L.; Benkhelifa-Ziyyat, S.; Rabai, A.; Mamchaoui, K.; Ferry, A.; et al. Allele-specific silencing therapy for Dynamin 2-related dominant centronuclear myopathy. EMBO Mol. Med. 2018, 10, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Shimizu, W.; Wilde, A.A.; Towbin, J.A.; Zareba, W.; Robinson, J.L.; Qi, M.; Vincent, G.M.; Ackerman, M.J.; Kaufman, E.S.; et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 2007, 115, 2481–2489. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Wei, H.; Lu, J.; Huang, D.; Liu, Z.; Loh, L.J.; Islam, O.; Liew, R.; Shim, W.; Cook, S.A. Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2015, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Takaki, T.; Inagaki, A.; Chonabayashi, K.; Inoue, K.; Miki, K.; Ohno, S.; Makiyama, T.; Horie, M.; Yoshida, Y. Optical recording of action potentials in human induced pluripotent stem cell-derived cardiac single cells and monolayers generated from long QT syndrome type 1 patients. Stem Cells Int. 2019, 2019, 7532657. [Google Scholar] [CrossRef]
- Sala, L.; Bellin, M.; Mummery, C.L. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: Has the time come? Br. J. Pharmacol. 2017, 174, 3749–3765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuriyanghai, Y.; Makiyama, T.; Sasaki, K.; Kamakura, T.; Yamamoto, Y.; Hayano, M.; Harita, T.; Nishiuchi, S.; Chen, J.; Kohjitani, H.; et al. Complex aberrant splicing in the induced pluripotent stem cell-derived cardiomyocytes from a patient with long QT syndrome carrying KCNQ1-A344Aspl mutation. Heart Rhythm 2018, 15, 1566–1574. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R. Dynamic clamp in electrophysiological studies on stem cell-derived cardiomyocytes—Why and how? J. Cardiovasc. Pharmacol. 2021, 77, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.X.; Di Diego, J.M.; Goodrow, R.J.; Wu, Y.; Cordeiro, J.M.; Nesterenko, V.V.; Barajas-Martínez, H.; Hu, D.; Urrutia, J.; Desai, M.; et al. Maximum diastolic potential of human induced pluripotent stem cell-derived cardiomyocytes depends critically on IKr. PLoS ONE 2012, 7, e40288. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2006–H2017. [Google Scholar] [CrossRef] [PubMed]
- Vincent, G.M.; Timothy, K.W.; Leppert, M.; Keating, M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N. Engl. J. Med. 1992, 327, 846–852. [Google Scholar] [CrossRef]
- Harrison, S.M.; Riggs, E.R.; Maglott, D.R.; Lee, J.M.; Azzariti, D.R.; Niehaus, A.; Ramos, E.M.; Martin, C.L.; Landrum, M.J.; Rehm, H.L. Using ClinVar as a resource to support variant interpretation. Curr. Protoc. Hum. Genet. 2016, 89, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, N.; Zhang, L.; Huang, H.; Chen, Y.; Zheng, J.; Zhou, X.; Yi, F.; Du, Q.; Liang, Z. siRNA has greatly elevated mismatch tolerance at 3′-UTR sites. PLoS ONE 2012, 7, e49309. [Google Scholar] [CrossRef] [PubMed]
- Pfister, E.L.; Kennington, L.; Straubhaar, J.; Wagh, S.; Liu, W.; DiFiglia, M.; Landwehrmeyer, B.; Vonsattel, J.P.; Zamore, P.D.; Aronin, N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr. Biol. 2009, 19, 774–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, A.; Dirven, R.J.; Boender, J.; Atiq, F.; Anvar, S.Y.; Leebeek, F.W.G.; van Vlijmen, B.J.M.; Eikenboom, J. Ex vivo improvement of a von Willebrand disease type 2A phenotype using an allele-specific small-interfering RNA. Thromb. Haemost. 2020, 120, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Li, J.; Zhang, J.; Xu, C.; Jiang, Y.; Wu, Z.; Zhao, F.; Liao, L.; Chen, J.; Lin, Y.; et al. Comprehensive characterization of human genome variation by high coverage whole-genome sequencing of forty four Caucasians. PLoS ONE 2013, 8, e59494. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, R.; Hegele, R.A. Complex effects of laminopathy mutations on nuclear structure and function. Clin. Genet. 2019, 95, 199–209. [Google Scholar] [CrossRef]
- Lennermann, D.; Backs, J.; van den Hoogenhof, M.M.G. New insights in RBM20 cardiomyopathy. Curr. Heart. Fail Rep. 2020, 17, 234–246. [Google Scholar] [CrossRef]
- Boink, G.J.; Robinson, R.B. Gene therapy for restoring heart rhythm. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Lorenzer, C.; Dirin, M.; Winkler, A.M.; Baumann, V.; Winkler, J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J Control Release 2015, 203, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.M.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef]
- Boukens, B.J.; Walton, R.; Meijborg, V.M.; Coronel, R. Transmural electrophysiological heterogeneity, the T-wave and ventricular arrhythmias. Prog. Biophys. Mol. Biol. 2016, 122, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J. Am. Coll Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tijsen, A.J.; Cócera Ortega, L.; Reckman, Y.J.; Zhang, X.; van der Made, I.; Aufiero, S.; Li, J.; Kamps, S.C.; van den Bout, A.; Devalla, H.D.; et al. Titin circular RNAs create a back-splice motif essential for SRSF10 splicing. Circulation 2021, 143, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Shinnawi, R.; Huber, I.; Maizels, L.; Shaheen, N.; Gepstein, A.; Arbel, G.; Tijsen, A.J.; Gepstein, L. Monitoring human-induced pluripotent stem cell-derived cardiomyocytes with genetically encoded calcium and voltage fluorescent reporters. Stem Cell Rep. 2015, 5, 582–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ten Tusscher, K.H.; Noble, D.; Noble, P.J.; Panfilov, A.V. A model for human ventricular tissue. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1573–H1589. [Google Scholar] [CrossRef] [PubMed]
- ten Tusscher, K.H.; Panfilov, A.V. Alternans and spiral breakup in a human ventricular tissue model. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1088–H1100. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, J.M.; Thygesen, H.H.; Schoneveld, O.J.; Das, A.T.; Berkhout, B.; Lamers, W.H. Factor correction as a tool to eliminate between-session variation in replicate experiments: Application to molecular biology and retrovirology. Retrovirology 2006, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cócera-Ortega, L.; Wilders, R.; Kamps, S.C.; Fabrizi, B.; Huber, I.; van der Made, I.; van den Bout, A.; de Vries, D.K.; Gepstein, L.; Verkerk, A.O.; et al. shRNAs Targeting a Common KCNQ1 Variant Could Alleviate Long-QT1 Disease Severity by Inhibiting a Mutant Allele. Int. J. Mol. Sci. 2022, 23, 4053. https://doi.org/10.3390/ijms23074053
Cócera-Ortega L, Wilders R, Kamps SC, Fabrizi B, Huber I, van der Made I, van den Bout A, de Vries DK, Gepstein L, Verkerk AO, et al. shRNAs Targeting a Common KCNQ1 Variant Could Alleviate Long-QT1 Disease Severity by Inhibiting a Mutant Allele. International Journal of Molecular Sciences. 2022; 23(7):4053. https://doi.org/10.3390/ijms23074053
Chicago/Turabian StyleCócera-Ortega, Lucía, Ronald Wilders, Selina C. Kamps, Benedetta Fabrizi, Irit Huber, Ingeborg van der Made, Anouk van den Bout, Dylan K. de Vries, Lior Gepstein, Arie O. Verkerk, and et al. 2022. "shRNAs Targeting a Common KCNQ1 Variant Could Alleviate Long-QT1 Disease Severity by Inhibiting a Mutant Allele" International Journal of Molecular Sciences 23, no. 7: 4053. https://doi.org/10.3390/ijms23074053