
Overcoming Steroid Resistance in Pediatric Acute Lymphoblastic Leukemia—The State-of-the-Art Knowledge and Future Prospects

 ,
,
Abstract
:1. Introduction
2. Concepts of Glucocorticoid Resistance in Pediatric ALL
2.1. Activation of Glucocorticoid Receptor—First Things First
2.2. Bcl-2 Protein Family as a Critical Mediator of Glucocorticoid-Induced Apoptosis
2.3. Role of Proteasomal Degradation in Resistance to Glucocorticoid Treatment in Pediatric ALL
2.4. IKZF1 Alterations and Glucocorticoid Resistance
3. Signaling Pathways Contribute to Glucocorticoid Resistance in Pediatric ALL—Prospects for Future Treatment
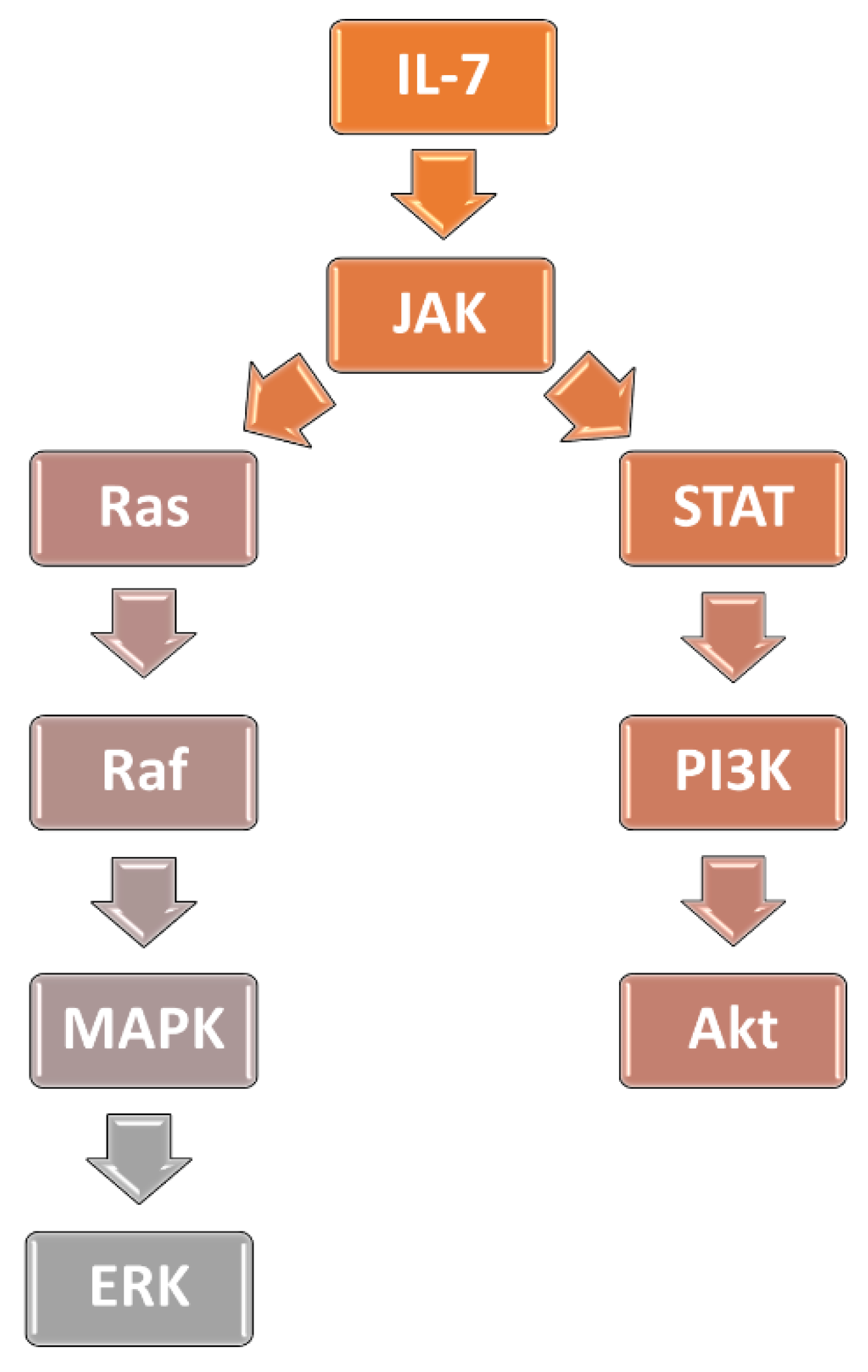
3.1. Interleukin-7 Signaling Pathway and Glucocorticoid Resistance in ALL
3.2. Activation of PI3K/AKT/mTOR Signaling Cascade Prevents GR from Translocation to the Nucleus
3.3. The MAPK-ERK Pathway
4. Methods of Enhancing the Results of Glucocorticoid Therapy in Pediatric ALL
4.1. Enhancing Effects of GR Activation
4.2. The BH3 Mimetics—Targeting the Primary Mechanism of Glucocorticoid-Induced Apoptosis of ALL Cells
4.3. Proteasome Inhibitors—Multitude Mechanism of Action Benefiting the Glucocorticoid Treatment
5. Other Prospects of Enhancing Glucocorticoid Sensitivity in ALL
5.1. Tigecycline
5.2. Tamoxifen
5.3. Cannabidiol
5.4. Mebendazole
5.5. Demethylating Agents
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| AIEOP-BFM ALL | Associazione Italiana di Ematologia Oncologia Pediatrica-Berlin-Frankfurt-Münster Acute Lymphoblastic Leukemia |
| AKT | serine/threonine kinase |
| AML | acute myeloid leukemia |
| ATF4 | cyclic AMP-dependent transcription factor |
| Bad | Bcl-2-associated agonist of cell death |
| Bak | Bcl-2 homologous antagonist/killer |
| Bax | Bcl-2-associated X-protein 4 |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-w | B-cell lymphoma-w |
| Bcl-XL | B-cell lymphoma extra-large |
| Bid | BH3 interacting-domain death agonist |
| BIM | Bcl-2-interacting mediator of cell death |
| cAMP-PKA | cyclic adenosine monophosphate-dependent protein kinase |
| CASP1 | caspaze 1 |
| CBD | cannabidiol |
| CHOP | C/EBP Homologous Protein |
| CI | combination index |
| CNS | central nervous system |
| CR | complete response |
| CRLF2 | cytokine receptor-like factor 2 |
| dmPGE2 | 6,16-dimethyl-prostaglandin E2 |
| DN | double negative |
| DR | death receptor |
| EFS | event-free survival |
| EMA | European Medicines Agency |
| ER | extreme risk |
| ERK | extracellular signal-regulated kinase |
| ESRRB | orphan nuclear receptor estrogen-related receptor-b |
| ETP | early T-cell progenitor |
| FBXW7 | F-Box and WD Repeat Domain-Containing 7 |
| FDA | Food and Drug Administration |
| FKBP51 | FK506-binding protein 51 |
| FOXO3a | forkhead box O3 |
| GR | glucocorticoid receptor |
| GRE | glucocorticoid response element |
| GSK3 | glycogen synthase kinase-3 |
| GTG | glucocorticoid target gene |
| HR | high risk |
| Hrk | activator of apoptosis harakiri |
| HyperCVAD | hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone |
| IC50 | half maximal inhibitory concentration |
| IGR | intronic GR binding region |
| IL-6 | interleukin 6 |
| IL-7 | interleukin-7 |
| IL-7R | IL-7 receptor |
| JAK | Janus kinases |
| LC50 | median lethal concentration |
| MAPK | mitogen-activated protein kinase |
| Mcl-1 | induced myeloid leukemia cell differentiation protein |
| miRNA | microRNA |
| MLL | mixed lineage leukemia |
| MRD | minimal residual disease |
| mRNA | messenger RNA |
| mTOR | mammalian target of rapamycin kinase |
| NF-κB | nuclear factor-κB |
| NOD | nonobese diabetic |
| Notch | neurogenic locus notch homolog protein |
| Noxa | phorbol-12-myristate-13-acetate-induced protein 1 |
| OXPHOS | oxidative phosphorylation |
| PERK | protein kinase R -like endoplasmic reticulum kinase |
| PGR | prednisolone good response |
| Ph-like | Philadelphia chromosome-like |
| PI3K | phosphatidylinositol 3-kinase |
| PKR | protein kinase R |
| PPR | prednisolone poor response |
| pre-B ALL | precursor-B-cell ALL |
| Puma | p53-upregulated modulator of apoptosis |
| Raf | rapidly accelerated fibrosarcoma |
| Ras | rat sarcoma virus |
| RER | endoplasmic reticulum |
| ROS | reactive oxygen species |
| RT-PCR | real-time polymerase chain reaction |
| SCID | severe combined immunodeficiency |
| SE | survival estimates |
| shRNA | short hairpin RNA |
| SIRT1 | sirtuin 1 |
| SR | standard risk |
| STAT | signal transducer and activator of transcription |
| T-ALL | precursor T-cell ALL |
| T-LL | T-lymphoblastic lymphoma |
| TNF | tumor-necrosis-factor |
| TRB3 | tribbles homolog 3 |
| VDAC | voltage-dependent anion channel 1 |
| VXL | vincristine, dexamethasone and L-asparaginase |
| γc | γ chain |
| Δ9-THC | Δ9-tetrahydrocannabinol |
References
- Stiller, C.A. Epidemiology and Genetics of Childhood Cancer. Oncogene 2004, 23, 6429–6444. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.S.; Kumar, R. Steroid Resistance in Leukemia. World J. Exp. Med. 2013, 3, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved Survival for Children and Adolescents With Acute Lymphoblastic Leukemia Between 1990 and 2005: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; de Rossi, G.; et al. Molecular Response to Treatment Redefines All Prognostic Factors in Children and Adolescents with B-Cell Precursor Acute Lymphoblastic Leukemia: Results in 3184 Patients of the AIEOP-BFM ALL 2000 Study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S. P Glucocorticoid Selection for Pediatric ALL. Blood 2016, 127, 2049–2051. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Pui, C.-H. Glucocorticoid Use in Acute Lymphoblastic Leukaemia. Lancet Oncol. 2010, 11, 1096–1106. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liu, W.J. Prognostic Value of the Response to Prednisone for Children with Acute Lymphoblastic Leukemia: A Meta-Analysis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7858–7866. [Google Scholar] [CrossRef]
- van der Zwet, J.C.G.; Smits, W.; Buijs-Gladdines, J.G.C.A.M.; Pieters, R.; Meijerink, J.P.P. Recurrent NR3C1 Aberrations at First Diagnosis Relate to Steroid Resistance in Pediatric T-Cell Acute Lymphoblastic Leukemia Patients. HemaSphere 2020, 5, e513. [Google Scholar] [CrossRef]
- Caratti, G.; Matthews, L.; Poolman, T.; Kershaw, S.; Baxter, M.; Ray, D. Glucocorticoid Receptor Function in Health and Disease. Clin. Endocrinol. 2015, 83, 441–448. [Google Scholar] [CrossRef]
- Vettorazzi, S.; Nalbantoglu, D.; Gebhardt, J.C.M.; Tuckermann, J. A Guide to Changing Paradigms of Glucocorticoid Receptor Function—A Model System for Genome Regulation and Physiology. FEBS J. 2021, 2, febs.16100. [Google Scholar] [CrossRef]
- Nick, Z.L.U.; Cidlowski, J.A. The Origin and Functions of Multiple Human Glucocorticoid Receptor Isoforms. Ann. N. Y. Acad. Sci. 2004, 1024, 102–123. [Google Scholar] [CrossRef]
- Lu, N.Z.; Cidlowski, J.A. Translational Regulatory Mechanisms Generate N-Terminal Glucocorticoid Receptor Isoforms with Unique Transcriptional Target Genes. Mol. Cell 2005, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Specificity and Sensitivity of Glucocorticoid Signaling in Health and Disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, J.C.; Oakley, R.H.; Jewell, C.M.; Cidlowski, J.A. Proinflammatory Cytokines Regulate Human Glucocorticoid Receptor Gene Expression and Lead to the Accumulation of the Dominant Negative Beta Isoform: A Mechanism for the Generation of Glucocorticoid Resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 6865–6870. [Google Scholar] [CrossRef] [Green Version]
- Giordano, P.; Molinari, A.C.; del Vecchio, G.C.; Saracco, P.; Russo, G.; Altomare, M.; Perutelli, P.; Crescenzio, N.; Santoro, N.; Marchetti, M.; et al. Prospective Study of Hemostatic Alterations in Children with Acute Lymphoblastic Leukemia. Am. J. Hematol. 2010, 85, 325–330. [Google Scholar] [CrossRef]
- Beger, C.; Gerdes, K.; Lauten, M.; Tissing, W.J.E.; Fernandez-Munoz, I.; Schrappe, M.; Welte, K. Expression and Structural Analysis of Glucocorticoid Receptor Isoform Gamma in Human Leukaemia Cells Using an Isoform-Specific Real-Time Polymerase Chain Reaction Approach. Br. J. Haematol. 2003, 122, 245–252. [Google Scholar] [CrossRef]
- Kuster, L.; Grausenburger, R.; Fuka, G.; Kaindl, U.; Krapf, G.; Inthal, A.; Mann, G.; Kauer, M.; Rainer, J.; Kofler, R.; et al. ETV6/RUNX1-Positive Relapses Evolve from an Ancestral Clone and Frequently Acquire Deletions of Genes Implicated in Glucocorticoid Signaling. Blood 2011, 117, 2658–2667. [Google Scholar] [CrossRef]
- Bokemeyer, A.; Eckert, C.; Meyr, F.; Koerner, G.; von Stackelberg, A.; Ullmann, R.; Türkmen, S.; Henze, G.; Seeger, K. Copy Number Genome Alterations Are Associated with Treatment Response and Outcome in Relapsed Childhood ETV6/RUNX1-Positive Acute Lymphoblastic Leukemia. Haematologica 2014, 99, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Grausenburger, R.; Bastelberger, S.; Eckert, C.; Kauer, M.; Stanulla, M.; Frech, C.; Bauer, E.; Stoiber, D.; von Stackelberg, A.; Attarbaschi, A.; et al. Genetic Alterations in Glucocorticoid Signaling Pathway Components Are Associated with Adverse Prognosis in Children with Relapsed ETV6/RUNX1-Positive Acute Lymphoblastic Leukemia. Leuk. Lymphoma 2016, 57, 1163–1173. [Google Scholar] [CrossRef]
- Sun, C.; Chang, L.; Zhu, X. Pathogenesis of ETV6/RUNX1-Positive Childhood Acute Lymphoblastic Leukemia and Mechanisms Underlying Its Relapse. Oncotarget 2017, 8, 35445–35459. [Google Scholar] [CrossRef] [Green Version]
- Irving, J.A.E.; Enshaei, A.; Parker, C.A.; Sutton, R.; Kuiper, R.P.; Erhorn, A.; Minto, L.; Venn, N.C.; Law, T.; Yu, J.; et al. Integration of Genetic and Clinical Risk Factors Improves Prognostication in Relapsed Childhood B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 2016, 128, 911–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Li, Z.; Qiu, F.; Li, C.; Lin, X.; He, Y.; Qian, M.; Song, Y.; Zhang, H. Association Between NR3C1 Mutations and Glucocorticoid Resistance in Children With Acute Lymphoblastic Leukemia. Front. Pharmacol. 2021, 12, 634956. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Ding, Y.; Gao, Y.; Wang, L.M.; Wang, H.; Ding, L.; Li, X.; Yu, X.; Huang, H. Haploinsufficiency of NR3C1 Drives Glucocorticoid Resistance in Adult Acute Lymphoblastic Leukemia Cells by Down-Regulating the Mitochondrial Apoptosis Axis, and Is Sensitive to Bcl-2 Blockage. Cancer Cell Int. 2019, 19, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, M.D.; Wolf, I.M.; Sanchez, E.R.; Witchel, S.F.; DeFranco, D.B. Glucocorticoid Receptor Physiology. Rev. Endocr. Metab. Disord. 2007, 8, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Schoneveld, O.J.L.M.; Gaemers, I.C.; Lamers, W.H. Mechanisms of Glucocorticoid Signalling. Biochim. Biophys. Acta 2004, 1680, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; van Vlierberghe, P. Overcoming Steroid Resistance in T Cell Acute Lymphoblastic Leukemia. PLoS Med. 2016, 13, e1002208. [Google Scholar] [CrossRef] [Green Version]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis Initiated When BH3 Ligands Engage Multiple Bcl-2 Homologs, Not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef] [Green Version]
- Gavathiotis, E.; Suzuki, M.; Davis, M.L.; Pitter, K.; Bird, G.H.; Katz, S.G.; Tu, H.C.; Kim, H.; Cheng, E.H.Y.; Tjandra, N.; et al. BAX Activation Is Initiated at a Novel Interaction Site. Nature 2008, 455, 1076–1081. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.D.; Cheng, E.H.Y. Hierarchical Regulation of Mitochondrion-Dependent Apoptosis by BCL-2 Subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358. [Google Scholar] [CrossRef]
- Cartron, P.F.; Gallenne, T.; Bougras, G.; Gautier, F.; Manero, F.; Vusio, P.; Meflah, K.; Vallette, F.M.; Juin, P. The First Alpha Helix of Bax Plays a Necessary Role in Its Ligand-Induced Activation by the BH3-Only Proteins Bid and PUMA. Mol. Cell 2004, 16, 807–818. [Google Scholar] [CrossRef]
- Harada, H.; Quearry, B.; Ruiz-Vela, A.; Korsmeyer, S.J. Survival Factor-Induced Extracellular Signal-Regulated Kinase Phosphorylates BIM, Inhibiting Its Association with BAX and Proapoptotic Activity. Proc. Natl. Acad. Sci. USA 2004, 101, 15313–15317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 Domains of BH3-Only Proteins Differentially Regulate Bax-Mediated Mitochondrial Membrane Permeabilization Both Directly and Indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.R.; Newmeyer, D.D. Bid, Bax, and Lipids Cooperate to Form Supramolecular Openings in the Outer Mitochondrial Membrane. Cell 2002, 111, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Marani, M.; Tenev, T.; Hancock, D.; Downward, J.; Lemoine, N.R. Identification of Novel Isoforms of the BH3 Domain Protein Bim Which Directly Activate Bax to Trigger Apoptosis. Mol. Cell. Biol. 2002, 22, 3577–3589. [Google Scholar] [CrossRef] [Green Version]
- Walensky, L.D.; Pitter, K.; Morash, J.; Oh, K.J.; Barbuto, S.; Fisher, J.; Smith, E.; Verdine, G.L.; Korsmeyer, S.J. A Stapled BID BH3 Helix Directly Binds and Activates BAX. Mol. Cell 2006, 24, 199–210. [Google Scholar] [CrossRef]
- Maung, Z.T.; MacLean, F.R.; Reid, M.M.; Pearson, A.D.J.; Proctor, S.J.; Hamilton, P.J.; Hall, A.G. The Relationship between Bcl-2 Expression and Response to Chemotherapy in Acute Leukaemia. Br. J. Haematol. 1994, 88, 105–109. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Kitanaka, A.; Pui, C.H.; McNinch, L.; Evans, W.E.; Raimondi, S.C.; Behm, F.G.; Aricò, M.; Campana, D. Clinical Relevance of BCL-2 Overexpression in Childhood Acute Lymphoblastic Leukemia. Blood 1996, 87, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Klobusická, M.; Kusenda, J.; Babusíkova, O. Expression of P53 and Bcl-2 Proteins in Acute Leukemias: An Immunocytochemical Study. Neoplasma 2001, 48, 489–495. [Google Scholar]
- Brown, L.M.; Hanna, D.T.; Khaw, S.L.; Ekert, P.G. Dysregulation of BCL-2 Family Proteins by Leukemia Fusion Genes. J. Biol. Chem. 2017, 292, 14325. [Google Scholar] [CrossRef] [Green Version]
- Prokop, A.; Wieder, T.; Sturm, I.; Emann, F.; Seeger, K.; Wuchter, C.; Ludwig, W.D.; Henze, G.; Dörken, B.; Daniel, P.T. Relapse in Childhood Acute Lymphoblastic Leukemia Is Associated with a Decrease of the Bax/Bcl-2 Ratio and Loss of Spontaneous Caspase-3 Processing in Vivo. Leukemia 2000, 14, 1606–1613. [Google Scholar] [CrossRef] [Green Version]
- Wojcik, I.; Szybka, M.; Golanska, E.; Rieske, P.; Blonski, J.Z.; Robak, T.; Bartkowiak, J. Abnormalities of the P53, MDM2, BCL2 and BAX genes in acute leukemias. Neoplasma 2005, 52, 318–324. [Google Scholar] [PubMed]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene Expression Signatures Define Novel Oncogenic Pathways in T Cell Acute Lymphoblastic Leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Sarang, Z.; Gyurina, K.; Scholtz, B.; Kiss, C.; Szegedi, I. Altered Expression of Autophagy-Related Genes Might Contribute to Glucocorticoid Resistance in Precursor B-Cell-Type Acute Lymphoblastic Leukemia. Eur. J. Haematol. 2016, 97, 453–460. [Google Scholar] [CrossRef]
- Wang, Z.; Malone, M.H.; He, H.; McColl, K.S.; Distelhorst, C.W. Microarray Analysis Uncovers the Induction of the Proapoptotic BH3-Only Protein Bim in Multiple Models of Glucocorticoid-Induced Apoptosis. J. Biol. Chem. 2003, 278, 23861–23867. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.N.; Guo, X.; Ma, Z.G.; Gu, L.; Ge, J.; Li, Q. Pro-Apoptotic Protein BIM in Apoptosis of Glucocorticoid-Sensitive and -Resistant Acute Lymphoblastic Leukemia CEM Cells. Med. Oncol. 2011, 28, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Abrams, M.T.; Robertson, N.M.; Yoon, K.; Wickstrom, E. Inhibition of Glucocorticoid-Induced Apoptosis by Targeting the Major Splice Variants of BIM MRNA with Small Interfering RNA and Short Hairpin RNA. J. Biol. Chem. 2004, 279, 55809–55817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlacher, M.; Michalak, E.M.; Kelly, P.N.; Labi, V.; Niederegger, H.; Coultas, L.; Adams, J.M.; Strasser, A.; Villunger, A. BH3-Only Proteins Puma and Bim Are Rate-Limiting for Gamma-Radiation- and Glucocorticoid-Induced Apoptosis of Lymphoid Cells in Vivo. Blood 2005, 106, 4131–4138. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, P.S.; Gorman, R.; MacKenzie, K.L.; Lutze-Mann, L.; Lock, R.B. Dexamethasone Resistance in B-Cell Precursor Childhood Acute Lymphoblastic Leukemia Occurs Downstream of Ligand-Induced Nuclear Translocation of the Glucocorticoid Receptor. Blood 2005, 105, 2519–2526. [Google Scholar] [CrossRef]
- Schmidt, S.; Rainer, J.; Riml, S.; Ploner, C.; Jesacher, S.; Achmüller, C.; Presul, E.; Skvortsov, S.; Crazzolara, R.; Fiegl, M.; et al. Identification of Glucocorticoid-Response Genes in Children with Acute Lymphoblastic Leukemia. Blood 2006, 107, 2061–2069. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates with c-Myc to Immortalize Pre-B Cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Laane, E.; Panaretakis, T.; Pokrovskaja, K.; Buentke, E.; Corcoran, M.; Söderhäll, S.; Heyman, M.; Mazur, J.; Zhivotovsky, B.; Porwit, A.; et al. Dexamethasone-Induced Apoptosis in Acute Lymphoblastic Leukemia Involves Differential Regulation of Bcl-2 Family Members. Haematologica 2007, 92, 1460–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, D.; Bhadri, V.A.; Beck, D.; Thoms, J.A.I.; Yakob, N.A.; Wong, J.W.H.; Knezevic, K.; Pimanda, J.E.; Lock, R.B. Opposing Regulation of BIM and BCL2 Controls Glucocorticoid-Induced Apoptosis of Pediatric Acute Lymphoblastic Leukemia Cells. Blood 2015, 125, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asnafi, V.; Buzyn, A.; le Noir, S.; Baleydier, F.; Simon, A.; Beldjord, K.; Reman, O.; Witz, F.; Fagot, T.; Tavernier, E.; et al. NOTCH1/FBXW7 Mutation Identifies a Large Subgroup with Favorable Outcome in Adult T-Cell Acute Lymphoblastic Leukemia (T-ALL): A Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) Study. Blood 2009, 113, 3918–3924. [Google Scholar] [CrossRef] [PubMed]
- Abdelali, R.B.; Asnafi, V.; Leguay, T.; Boissel, N.; Buzyn, A.; Chevallier, P.; Thomas, X.; Lepretre, S.; Huguet, F.; Vey, N.; et al. Pediatric-Inspired Intensified Therapy of Adult T-ALL Reveals the Favorable Outcome of NOTCH1/FBXW7 Mutations, but Not of Low ERG/BAALC Expression: A GRAALL Study. Blood 2011, 118, 5099–5107. [Google Scholar] [CrossRef] [Green Version]
- Breit, S.; Stanulla, M.; Flohr, T.; Schrappe, M.; Ludwig, W.D.; Tolle, G.; Happich, M.; Muckenthaler, M.U.; Kulozik, A.E. Activating NOTCH1 Mutations Predict Favorable Early Treatment Response and Long-Term Outcome in Childhood Precursor T-Cell Lymphoblastic Leukemia. Blood 2006, 108, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Clappier, E.; Collette, S.; Grardel, N.; Girard, S.; Suarez, L.; Brunie, G.; Kaltenbach, S.; Yakouben, K.; Mazingue, F.; Robert, A.; et al. NOTCH1 and FBXW7 Mutations Have a Favorable Impact on Early Response to Treatment, but Not on Outcome, in Children with T-Cell Acute Lymphoblastic Leukemia (T-ALL) Treated on EORTC Trials 58881 and 58951. Leukemia 2010, 24, 2023–2031. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.J. Parvovirus-Induced Regression of Canine Transmissible Venereal Sarcoma. Am. J. Vet. Res. 1987, 48, 799–800. [Google Scholar]
- Malyukova, A.; Dohda, T.; von der Lehr, N.; Akhondi, S.; Corcoran, M.; Heyman, M.; Spruck, C.; Grandér, D.; Lendahl, U.; Sangfelt, O. The Tumor Suppressor Gene HCDC4 Is Frequently Mutated in Human T-Cell Acute Lymphoblastic Leukemia with Functional Consequences for Notch Signaling. Cancer Res. 2007, 67, 5611–5616. [Google Scholar] [CrossRef] [Green Version]
- Park, M.J.; Taki, T.; Oda, M.; Watanabe, T.; Yumura-Yagi, K.; Kobayashi, R.; Suzuki, N.; Hara, J.; Horibe, K.; Hayashi, Y. FBXW7 and NOTCH1 Mutations in Childhood T Cell Acute Lymphoblastic Leukaemia and T Cell Non-Hodgkin Lymphoma. Br. J. Haematol. 2009, 145, 198–206. [Google Scholar] [CrossRef]
- Malyukova, A.; Brown, S.; Papa, R.; O’Brien, R.; Giles, J.; Trahair, T.N.; Dalla Pozza, L.; Sutton, R.; Liu, T.; Haber, M.; et al. FBXW7 Regulates Glucocorticoid Response in T-Cell Acute Lymphoblastic Leukaemia by Targeting the Glucocorticoid Receptor for Degradation. Leukemia 2013, 27, 1053–1062. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, L.; Verhoog, N.J.D.; Louw, A. Disease- and Treatment-Associated Acquired Glucocorticoid Resistance. Endocr. Connect. 2018, 7, R328–R349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploner, C.; Rainer, J.; Lobenwein, S.; Geley, S.; Kofler, R. Repression of the BH3-Only Molecule PMAIP1/Noxa Impairs Glucocorticoid Sensitivity of Acute Lymphoblastic Leukemia Cells. Apoptosis 2009, 14, 821–828. [Google Scholar] [CrossRef]
- Georgopoulos, K.; Bigby, M.; Wang, J.H.; Molnar, A.; Wu, P.; Winandy, S.; Sharpe, A. The Ikaros Gene Is Required for the Development of All Lymphoid Lineages. Cell 1994, 79, 143–156. [Google Scholar] [CrossRef]
- Kastner, P.; Chan, S. Role of Ikaros in T-Cell Acute Lymphoblastic Leukemia. World J. Biol. Chem. 2011, 2, 108. [Google Scholar] [CrossRef] [PubMed]
- Joshi, I.; Yoshida, T.; Jena, N.; Qi, X.; Zhang, J.; van Etten, R.A.; Georgopoulos, K. Ikaros Mutation Confers Integrin-Dependent Pre-B Cell Survival and Progression to Acute Lymphoblastic Leukemia. Nat. Immunol. 2014, 15, 294. [Google Scholar] [CrossRef]
- Oliveira, V.C.; Lacerda, M.P.; Moraes, B.B.; Gomes, C.P.; Maricato, J.T.; Souza, O.F.; Schenkman, S.; Pesquero, J.B.; Moretti, N.S.; Rodrigues, C.A.; et al. Deregulation of Ikaros Expression in B-1 Cells: New Insights in the Malignant Transformation to Chronic Lymphocytic Leukemia. J. Leukoc. Biol. 2019, 106, 581–594. [Google Scholar] [CrossRef]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; te Kronnie, G.; Bourquin, J.P.; Bornhauser, B.; et al. IKZF1 plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric b-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Med. 2018, 36, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Marke, R.; Havinga, J.; Cloos, J.; Demkes, M.; Poelmans, G.; Yuniati, L.; van Ingen Schenau, D.; Sonneveld, E.; Waanders, E.; Pieters, R.; et al. Tumor Suppressor IKZF1 Mediates Glucocorticoid Resistance in B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia 2015, 30, 1599–1603. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005. [Google Scholar] [CrossRef] [Green Version]
- Collins-Underwood, J.R.; Mullighan, C.G. Genomic Profiling of High-Risk Acute Lymphoblastic Leukemia. Leukemia 2010, 24, 1676–1685. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, R.P.; Waanders, E.; van der Velden, V.H.J.; van Reijmersdal, S.v.; Venkatachalam, R.; Scheijen, B.; Sonneveld, E.; van Dongen, J.J.M.; Veerman, A.J.P.; van Leeuwen, F.N.; et al. IKZF1 Deletions Predict Relapse in Uniformly Treated Pediatric Precursor B-ALL. Leukemia 2010, 24, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Cario, G.; Leoni, V.; Conter, V.; Baruchel, A.; Schrappe, M.; Biondi, A. BCR-ABL1-like Acute Lymphoblastic Leukemia in Childhood and Targeted Therapy. Haematologica 2020, 105, 2200. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Yano, M.; Asai, D.; Moriya-Saito, A.; Suenobu, S.I.; Hasegawa, D.; Deguchi, T.; Hashii, Y.; Kawasaki, H.; Hori, H.; et al. IKZF1 Deletion Is Enriched in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia Patients Showing Prednisolone Resistance. Leukemia 2016, 30, 1801–1803. [Google Scholar] [CrossRef]
- Braun, M.; Pastorczak, A.; Sędek, Ł.; Taha, J.; Madzio, J.; Jatczak-Pawlik, I.; Wypyszczak, K.; Matysiak, M.; Derwich, K.; Lejman, M.; et al. Prognostic Significance of IKZF1 Deletions and IKZF1 plus Profile in Children with B-Cell Precursor Acute Lymphoblastic Leukemia Treated According to the ALL-IC BFM 2009 Protocol. Hematol. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Grabstein, K.H.; Waldschmidt, T.J.; Finkelman, F.D.; Hess, B.W.; Alpert, A.R.; Boiani, N.E.; Namen, A.E.; Morrissey, P.J. Inhibition of Murine B and T Lymphopoiesis in Vivo by an Anti-Interleukin 7 Monoclonal Antibody. J. Exp. Med. 1993, 178, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Sudo, T.; Nishikawa, S.; Ohno, N.; Akiyama, N.; Tamakoshi, M.; Yoshida, H.; Nishikawa, S.I. Expression and Function of the Interleukin 7 Receptor in Murine Lymphocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 9125–9129. [Google Scholar] [CrossRef] [Green Version]
- Chazen, G.D.; Pereira, G.M.B.; LeGros, G.; Gillis, S.; Shevach, E.M. Interleukin 7 Is a T-Cell Growth Factor. Proc. Natl. Acad. Sci. USA 1989, 86, 5923–5927. [Google Scholar] [CrossRef] [Green Version]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrançois, L. Interleukin-7 Mediates the Homeostasis of Naïve and Memory CD8 T Cells in Vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef]
- Kittipatarin, C.; Khaled, A.R. Interlinking Interleukin-7. Cytokine 2007, 39, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Nakajima, H.; Saito, Y.; Saito, T.; Leonard, W.J.; Iwamoto, I. Janus Kinase 3 (Jak3) Is Essential for Common Cytokine Receptor Gamma Chain (Gamma(c))-Dependent Signaling: Comparative Analysis of Gamma(c), Jak3, and Gamma(c) and Jak3 Double-Deficient Mice. Int. Immunol. 2000, 12, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, D.; Melão, A.; van Boxtel, R.; Santos, C.I.; Silva, A.; Silva, M.C.; Cardoso, B.A.; Coffer, P.J.; Barata, J.T. STAT5 Is Essential for IL-7-Mediated Viability, Growth, and Proliferation of T-Cell Acute Lymphoblastic Leukemia Cells. Blood Adv. 2018, 2, 2199–2213. [Google Scholar] [CrossRef] [PubMed]
- Barata, J.T.; Cardoso, A.A.; Boussiotis, V.A. Interleukin-7 in T-Cell Acute Lymphoblastic Leukemia: An Extrinsic Factor Supporting Leukemogenesis? Leuk. Lymphoma 2005, 46, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R Gain-of-Function Mutations in Childhood T-Cell Acute Lymphoblastic Leukemia. Nat. Genet. 2011, 43, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.; Ma, M.C.; Snow, J.W.; Miners, M.J.; Herndier, B.G.; Goldsmith, M.A. Haploinsufficiency Identifies STAT5 as a Modifier of IL-7-Induced Lymphomas. Oncogene 2005, 24, 5252–5257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.; Laranjeira, A.B.A.; Martins, L.R.; Cardoso, B.A.; Demengeot, J.; Andrés Yunes, J.; Seddon, B.; Barata, J.T. IL-7 Contributes to the Progression of Human T-Cell Acute Lymphoblastic Leukemias. Cancer Res. 2011, 71, 4780–4789. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.; Schwab, C.; Broux, M.; Geerdens, E.; Degryse, S.; Demeyer, S.; Lahortiga, I.; Elliott, A.; Chilton, L.; la Starza, R.; et al. Targeted Sequencing Identifies Associations between IL7R-JAK Mutations and Epigenetic Modulators in T-Cell Acute Lymphoblastic Leukemia. Haematologica 2015, 100, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-Function Mutations in Interleukin-7 Receptor-α (IL7R) in Childhood Acute Lymphoblastic Leukemias. J. Exp. Med. 2011, 208, 901–908. [Google Scholar] [CrossRef]
- Barata, J.T.; Silva, A.; Brandao, J.G.; Nadler, L.M.; Cardoso, A.A.; Boussiotis, V.A. Activation of PI3K Is Indispensable for Interleukin 7-Mediated Viability, Proliferation, Glucose Use, and Growth of T Cell Acute Lymphoblastic Leukemia Cells. J. Exp. Med. 2004, 200, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Buijs-Gladdines, J.G.C.A.M.; Canté-Barrett, K.; Stubbs, A.P.; Vroegindeweij, E.M.; Smits, W.K.; van Marion, R.; Dinjens, W.N.M.; Horstmann, M.; Kuiper, R.P.; et al. IL-7 Receptor Mutations and Steroid Resistance in Pediatric T Cell Acute Lymphoblastic Leukemia: A Genome Sequencing Study. PLoS Med. 2016, 13, e1002200. [Google Scholar] [CrossRef]
- Delgado-Martin, C.; Meyer, L.K.; Huang, B.J.; Shimano, K.A.; Zinter, M.S.; Nguyen, J.V.; Smith, G.A.; Taunton, J.; Winter, S.S.; Roderick, J.R.; et al. JAK/STAT Pathway Inhibition Overcomes IL7-Induced Glucocorticoid Resistance in a Subset of Human T-Cell Acute Lymphoblastic Leukemias. Leukemia 2017, 31, 2568–2576. [Google Scholar] [CrossRef]
- Meyer, L.K.; Huang, B.J.; Delgado-Martin, C.; Roy, R.P.; Hechmer, A.; Wandler, A.M.; Vincent, T.L.; Fortina, P.; Olshen, A.B.; Wood, B.L.; et al. Glucocorticoids Paradoxically Facilitate Steroid Resistance in T Cell Acute Lymphoblastic Leukemias and Thymocytes. J. Clin. Investig. 2020, 130, 863–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barata, J.T.; Durum, S.K.; Seddon, B. Flip the Coin: IL-7 and IL-7R in Health and Disease. Nat. Immunol. 2019, 20, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Tasian, S.K.; Vincent, T.; Hall, J.W.; Sheen, C.; Roberts, K.G.; Seif, A.E.; Barrett, D.M.; Chen, I.M.; Collins, J.R.; et al. Targeting JAK1/2 and MTOR in Murine Xenograft Models of Ph-like Acute Lymphoblastic Leukemia. Blood 2012, 120, 3510–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, R.C.; Mullighan, C.G.; Chen, I.M.; Wharton, W.; Mikhail, F.M.; Carroll, A.J.; Kang, H.; Liu, W.; Dobbin, K.K.; Smith, M.A.; et al. Rearrangement of CRLF2 Is Associated with Mutation of JAK Kinases, Alteration of IKZF1, Hispanic/Latino Ethnicity, and a Poor Outcome in Pediatric B-Progenitor Acute Lymphoblastic Leukemia. Blood 2010, 115, 5312–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roll, J.D.; Reuther, G.W. CRLF2 and JAK2 in B-Progenitor Acute Lymphoblastic Leukemia: A Novel Association in Oncogenesis. Cancer Res. 2010, 70, 7347–7352. [Google Scholar] [CrossRef] [Green Version]
- Böhm, J.W.; Sia, K.C.S.; Jones, C.; Evans, K.; Mariana, A.; Pang, I.; Failes, T.; Zhong, L.; Mayoh, C.; Landman, R.; et al. Combination Efficacy of Ruxolitinib with Standard-of-Care Drugs in CRLF2-Rearranged Ph-like Acute Lymphoblastic Leukemia. Leukemia 2021, 35, 3101–3112. [Google Scholar] [CrossRef]
- Ellis, J.; van Maurik, A.; Fortunato, L.; Gisbert, S.; Chen, K.; Schwartz, A.; McHugh, S.; Want, A.; Santos Franco, S.; Oliveira, J.J.; et al. Anti-IL-7 Receptor α Monoclonal Antibody (GSK2618960) in Healthy Subjects—A Randomized, Double-Blind, Placebo-Controlled Study. Br. J. Clin. Pharmacol. 2019, 85, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Akkapeddi, P.; Fragoso, R.; Hixon, J.A.; Ramalho, A.S.; Oliveira, M.L.; Carvalho, T.; Gloger, A.; Matasci, M.; Corzana, F.; Durum, S.K.; et al. A Fully Human Anti-IL-7Rα Antibody Promotes Antitumor Activity against T-Cell Acute Lymphoblastic Leukemia. Leukemia 2019, 33, 2155–2168. [Google Scholar] [CrossRef]
- Piovan, E.; Yu, J.; Tosello, V.; Herranz, D.; Ambesi-Impiombato, A.; DaSilva, A.C.; Sanchez-Martin, M.; Perez-Garcia, A.; Rigo, I.; Castillo, M.; et al. Direct Reversal of Glucocorticoid Resistance by AKT Inhibition in Acute Lymphoblastic Leukemia. Cancer Cell 2013, 24, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Montaño, A.; Forero-Castro, M.; Marchena-Mendoza, D.; Benito, R.; Hernández-Rivas, J.M. New Challenges in Targeting Signaling Pathways in Acute Lymphoblastic Leukemia by NGS Approaches: An Update. Cancers 2018, 10, 110. [Google Scholar] [CrossRef] [Green Version]
- Schülein, C.; Eilers, M.; Popov, N. PI3K-Dependent Phosphorylation of Fbw7 Modulates Substrate Degradation and Activity. FEBS Lett. 2011, 585, 2151–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wandler, A.M.; Huang, B.J.; Craig, J.W.; Hayes, K.; Yan, H.; Meyer, L.K.; Scacchetti, A.; Monsalve, G.; Dail, M.; Li, Q.; et al. Loss of Glucocorticoid Receptor Expression Mediates in Vivo Dexamethasone Resistance in T-Cell Acute Lymphoblastic Leukemia. Leukemia 2020, 34, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Bressanin, D.; Evangelisti, C.; Ricci, F.; Tabellini, G.; Chiarini, F.; Tazzari, P.L.; Melchionda, F.; Buontempo, F.; Pagliaro, P.; Pession, A.; et al. Harnessing the PI3K/Akt/MTOR Pathway in T-Cell Acute Lymphoblastic Leukemia: Eliminating Activity by Targeting at Different Levels. Oncotarget 2012, 3, 811–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ksionda, O.; Mues, M.; Wandler, A.M.; Donker, L.; Tenhagen, M.; Jun, J.; Ducker, G.S.; Matlawska-Wasowska, K.; Shannon, K.; Shokat, K.M.; et al. Comprehensive Analysis of T Cell Leukemia Signals Reveals Heterogeneity in the PI3 Kinase-Akt Pathway and Limitations of PI3 Kinase Inhibitors as Monotherapy. PLoS ONE 2018, 13, e0193849. [Google Scholar] [CrossRef]
- Dail, M.; Wong, J.; Lawrence, J.; O’Connor, D.; Nakitandwe, J.; Chen, S.C.; Xu, J.; Lee, L.B.; Akagi, K.; Li, Q.; et al. Loss of Oncogenic Notch1 with Resistance to a PI3K Inhibitor in T-Cell Leukaemia. Nature 2014, 513, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Eldfors, S.; Kuusanmäki, H.; Kontro, M.; Majumder, M.M.; Parsons, A.; Edgren, H.; Pemovska, T.; Kallioniemi, O.; Wennerberg, K.; Gökbuget, N.; et al. Idelalisib Sensitivity and Mechanisms of Disease Progression in Relapsed TCF3-PBX1 Acute Lymphoblastic Leukemia. Leukemia 2017, 31, 51–57. [Google Scholar] [CrossRef]
- Katsuya, H.; Cook, L.B.M.; Rowan, A.G.; Satou, Y.; Taylor, G.P.; Bangham, C.R.M. Phosphatidylinositol 3-Kinase-δ (PI3K-δ) Is a Potential Therapeutic Target in Adult T-Cell Leukemia-Lymphoma. Biomark. Res. 2018, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.K.N.; Machado-Neto, J.A.; Lopes, M.R.; Morini, B.C.; Traina, F.; Costa, F.F.; Saad, S.T.O.; Favaro, P. Molecular Effects of the Phosphatidylinositol-3-Kinase Inhibitor NVP-BKM120 on T and B-Cell Acute Lymphoblastic Leukaemia. Eur. J. Cancer 2015, 51, 2076–2085. [Google Scholar] [CrossRef]
- Lonetti, A.; Antunes, I.L.; Chiarini, F.; Orsini, E.; Buontempo, F.; Ricci, F.; Tazzari, P.L.; Pagliaro, P.; Melchionda, F.; Pession, A.; et al. Activity of the Pan-Class I Phosphoinositide 3-Kinase Inhibitor NVP-BKM120 in T-Cell Acute Lymphoblastic Leukemia. Leukemia 2014, 28, 1196–1206. [Google Scholar] [CrossRef]
- Evangelisti, C.; Cappellini, A.; Oliveira, M.; Fragoso, R.; Barata, J.T.; Bertaina, A.; Locatelli, F.; Simioni, C.; Neri, L.M.; Chiarini, F.; et al. Phosphatidylinositol 3-Kinase Inhibition Potentiates Glucocorticoid Response in B-Cell Acute Lymphoblastic Leukemia. J. Cell. Physiol. 2018, 233, 1796–1811. [Google Scholar] [CrossRef]
- Teachey, D.T.; Obzut, D.A.; Cooperman, J.; Fang, J.; Carroll, M.; Choi, J.K.; Houghton, P.J.; Brown, V.I.; Grupp, S.A. The MTOR Inhibitor CCI-779 Induces Apoptosis and Inhibits Growth in Preclinical Models of Primary Adult Human ALL. Blood 2006, 107, 1149–1155. [Google Scholar] [CrossRef] [Green Version]
- Teachey, D.T.; Sheen, C.; Hall, J.; Ryan, T.; Brown, V.I.; Fish, J.; Reid, G.S.D.; Seif, A.E.; Norris, R.; Chang, Y.J.; et al. MTOR Inhibitors Are Synergistic with Methotrexate: An Effective Combination to Treat Acute Lymphoblastic Leukemia. Blood 2008, 112, 2020–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.; Welschinger, R.; Hewson, J.; Bradstock, K.F.; Bendall, L.J. Efficacy of Dual PI-3K and MTOR Inhibitors in Vitro and in Vivo in Acute Lymphoblastic Leukemia. Oncotarget 2014, 5, 10460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachmann, S.M.; Hofmann, I.; Schnell, C.; Fritsch, C.; Wee, S.; Lane, H.; Wang, S.; Garcia-Echeverria, C.; Maira, S.M. Specific Apoptosis Induction by the Dual PI3K/MTor Inhibitor NVP-BEZ235 in HER2 Amplified and PIK3CA Mutant Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 22299–22304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.; Wunderle, L.; Badura, S.; Schleyer, E.; Brüggemann, M.; Serve, H.; Schnittger, S.; Gökbuget, N.; Pfeifer, H.; Wagner, S.; et al. A Phase i Study of a Dual PI3-Kinase/MTOR Inhibitor BEZ235 in Adult Patients with Relapsed or Refractory Acute Leukemia. BMC Pharmacol. Toxicol. 2020, 21, 70. [Google Scholar] [CrossRef] [PubMed]
- Pongas, G.; Fojo, T.; Peter, J.J. BEZ235: When Promising Science Meets Clinical Reality. Oncologist 2016, 21, 1033–1034. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- van der Zwet, J.C.G.; Buijs-Gladdines, J.G.C.A.M.; Cordo’, V.; Debets, D.O.; Smits, W.K.; Chen, Z.; Dylus, J.; Zaman, G.J.R.; Altelaar, M.; Oshima, K.; et al. MAPK-ERK Is a Central Pathway in T-Cell Acute Lymphoblastic Leukemia That Drives Steroid Resistance. Leukemia 2021, 35, 3394–3405. [Google Scholar] [CrossRef]
- Khan, A.Q.; Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Dermime, S.; Uddin, S. RAS-Mediated Oncogenic Signaling Pathways in Human Malignancies. Semin. Cancer Biol. 2019, 54, 1–13. [Google Scholar] [CrossRef]
- Irving, J.; Matheson, E.; Minto, L.; Blair, H.; Case, M.; Halsey, C.; Swidenbank, I.; Ponthan, F.; Kirschner-Schwabe, R.; Groeneveld-Krentz, S.; et al. Ras Pathway Mutations Are Prevalent in Relapsed Childhood Acute Lymphoblastic Leukemia and Confer Sensitivity to MEK Inhibition. Blood 2014, 124, 3420–3430. [Google Scholar] [CrossRef]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; van de Ven, C.; de Groot-Kruseman, H.A.; de Haas, V.; et al. RAS Pathway Mutations as a Predictive Biomarker for Treatment Adaptation in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Matheson, E.C.; Thomas, H.; Case, M.; Blair, H.; Jackson, R.K.; Masic, D.; Veal, G.; Halsey, C.; Newell, D.R.; Vormoor, J.; et al. Glucocorticoids and Selumetinib Are Highly Synergistic in RAS Pathway-Mutated Childhood Acute Lymphoblastic Leukemia through Upregulation of BIM. Haematologica 2019, 104, 1804–1811. [Google Scholar] [CrossRef] [Green Version]
- Polak, A.; Kiliszek, P.; Sewastianik, T.; Szydłowski, M.; Jabłońska, E.; Białopiotrowicz, E.; Górniak, P.; Markowicz, S.; Nowak, E.; Grygorowicz, M.A.; et al. MEK Inhibition Sensitizes Precursor B-Cell Acute Lymphoblastic Leukemia (B-ALL) Cells to Dexamethasone through Modulation of MTOR Activity and Stimulation of Autophagy. PLoS ONE 2016, 11, e0155893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter-Pechańska, P.; Kunz, J.B.; Hof, J.; Zimmermann, M.; Rausch, T.; Bandapalli, O.R.; Orlova, E.; Scapinello, G.; Sagi, J.C.; Stanulla, M.; et al. Identification of a Genetically Defined Ultra-High-Risk Group in Relapsed Pediatric T-Lymphoblastic Leukemia. Blood Cancer J. 2017, 7, e523. [Google Scholar] [CrossRef] [Green Version]
- Oshima, K.; Khiabanian, H.; da Silva-Almeida, A.C.; Tzoneva, G.; Abate, F.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Carpenter, Z.; Penson, A.; Perez-Garcia, A.; et al. Mutational Landscape, Clonal Evolution Patterns, and Role of RAS Mutations in Relapsed Acute Lymphoblastic Leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, 11306–11311. [Google Scholar] [CrossRef] [Green Version]
- Gianfelici, V.; Chiaretti, S.; Demeyer, S.; di Giacomo, F.; Messina, M.; la Starza, R.; Peragine, N.; Paoloni, F.; Geerdens, E.; Pierini, V.; et al. RNA Sequencing Unravels the Genetics of Refractory/Relapsed T-Cell Acute Lymphoblastic Leukemia. Prognostic and Therapeutic Implications. Haematologica 2016, 101, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.; Trinquand, A.; Chevret, S.; Ballerini, P.; Cayuela, J.M.; Grardel, N.; Touzart, A.; Brethon, B.; Lapillonne, H.; Schmitt, C.; et al. Oncogenetic Mutations Combined with MRD Improve Outcome Prediction in Pediatric T-Cell Acute Lymphoblastic Leukemia. Blood 2018, 131, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Kerstjens, M.; Driessen, E.M.C.; Willekes, M.; Pinhanços, S.S.; Schneider, P.; Pieters, R.; Stam, R.W. MEK Inhibition Is a Promising Therapeutic Strategy for MLL-Rearranged Infant Acute Lymphoblastic Leukemia Patients Carrying RAS Mutations. Oncotarget 2017, 8, 14835–14846. [Google Scholar] [CrossRef] [Green Version]
- Driessen, E.M.C.; van Roon, E.H.J.; Spijkers-Hagelstein, J.A.P.; Schneider, P.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R.; Stam, R.W. Frequencies and Prognostic Impact of RAS Mutations in MLL-Rearranged Acute Lymphoblastic Leukemia in Infants. Haematologica 2013, 98, 937–944. [Google Scholar] [CrossRef]
- Pieters, R.; Schrappe, M.; de Lorenzo, P.; Hann, I.; de Rossi, G.; Felice, M.; Hovi, L.; LeBlanc, T.; Szczepanski, T.; Ferster, A.; et al. A Treatment Protocol for Infants Younger than 1 Year with Acute Lymphoblastic Leukaemia (Interfant-99): An Observational Study and a Multicentre Randomised Trial. Lancet 2007, 370, 240–250. [Google Scholar] [CrossRef]
- Degryse, S.; de Bock, C.E.; Demeyer, S.; Govaerts, I.; Bornschein, S.; Verbeke, D.; Jacobs, K.; Binos, S.; Skerrett-Byrne, D.A.; Murray, H.C.; et al. Mutant JAK3 Phosphoproteomic Profiling Predicts Synergism between JAK3 Inhibitors and MEK/BCL2 Inhibitors for the Treatment of T-Cell Acute Lymphoblastic Leukemia. Leukemia 2018, 32, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Schult, C.; Dahlhaus, M.; Glass, A.; Fischer, K.; Lange, S.; Freund, M.; Junghanss, C. The dual kinase inhibitor NVP-BEZ235 in combination with cytotoxic drugs exerts anti-proliferative activity towards acute lymphoblastic leukemia cells. Anticancer. Res. 2012, 32, 463–474. [Google Scholar] [PubMed]
- Tasian, S.K.; Assad, A.; Hunter, D.S.; Du, Y.; Loh, M.L. A Phase 2 Study of Ruxolitinib with Chemotherapy in Children with Philadelphia Chromosome-like Acute Lymphoblastic Leukemia (INCB18424-269/AALL1521): Dose-Finding Results from the Part 1 Safety Phase. Blood 2018, 132, 555. [Google Scholar] [CrossRef]
- Roderick, J.E.; Gallagher, K.M.; Murphy, L.C.; O’Connor, K.W.; Tang, K.; Zhang, B.; Brehm, M.A.; Greiner, D.L.; Yu, J.; Zhu, L.J.; et al. Prostaglandin E2 Stimulates CAMP Signaling and Resensitizes Human Leukemia Cells to Glucocorticoid-Induced Cell Death. Blood 2021, 137, 500–512. [Google Scholar] [CrossRef]
- Li, X.J.; Luo, X.Q.; Han, B.W.; Duan, F.T.; Wei, P.P.; Chen, Y.Q. MicroRNA-100/99a, Deregulated in Acute Lymphoblastic Leukaemia, Suppress Proliferation and Promote Apoptosis by Regulating the FKBP51 and IGF1R/MTOR Signalling Pathways. Br. J. Cancer 2013, 109, 2189–2198. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.N.; Tang, Y.L.; Ke, Z.Y.; Chen, Y.Q.; Luo, X.Q.; Zhang, H.; Huang, L.B. MiR-124 Contributes to Glucocorticoid Resistance in Acute Lymphoblastic Leukemia by Promoting Proliferation, Inhibiting Apoptosis and Targeting the Glucocorticoid Receptor. J. Steroid Biochem. Mol. Biol. 2017, 172, 62–68. [Google Scholar] [CrossRef]
- Paugh, S.W.; Bonten, E.J.; Savic, D.; Ramsey, L.B.; Thierfelder, W.E.; Gurung, P.; Malireddi, R.K.S.; Actis, M.; Mayasundari, A.; Min, J.; et al. NALP3 Inflammasome Upregulation and CASP1 Cleavage of the Glucocorticoid Receptor Cause Glucocorticoid Resistance in Leukemia Cells. Nat. Genet. 2015, 47, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, K.M.; Roderick, J.E.; Tan, S.H.; Tan, T.K.; Murphy, L.; Yu, J.; Li, R.; O’Connor, K.W.; Zhu, J.; Green, M.R.; et al. ESRRB Regulates Glucocorticoid Gene Expression in Mice and Patients with Acute Lymphoblastic Leukemia. Blood Adv. 2020, 4, 3154–3168. [Google Scholar] [CrossRef]
- Croce, C.M.; Reed, J.C. Finally, An Apoptosis-Targeting Therapeutic for Cancer. Cancer Res. 2016, 76, 5914–5920. [Google Scholar] [CrossRef] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Thol, F. What to Use to Treat AML: The Role of Emerging Therapies. Hematology 2021, 2021, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Held, L.; Siu, C.; Shadman, M. Venetoclax as a Therapeutic Option for the Treatment of Chronic Lymphocytic Leukemia: The Evidence so Far. Expert. Opin. Pharmacother. 2021, 22, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Ni Chonghaile, T.; Roderick, J.E.; Glenfield, C.; Ryan, J.; Sallan, S.E.; Silverman, L.B.; Loh, M.L.; Hunger, S.P.; Wood, B.; DeAngelo, D.J.; et al. Maturation Stage of T-Cell Acute Lymphoblastic Leukemia Determines BCL-2 versus BCL-XL Dependence and Sensitivity to ABT-199. Cancer Discov. 2014, 4, 1074–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peirs, S.; Matthijssens, F.; Goossens, S.; van de Walle, I.; Ruggero, K.; de Bock, C.E.; Degryse, S.; Canté-Barrett, K.; Briot, D.; Clappier, E.; et al. ABT-199 Mediated Inhibition of BCL-2 as a Novel Therapeutic Strategy in T-Cell Acute Lymphoblastic Leukemia. Blood 2014, 124, 3738–3747. [Google Scholar] [CrossRef]
- El-Cheikh, J.; Moukalled, N.M.; el Darsa, H.; Massoud, R.; Kanj, S.S.; Mahfouz, R.; Bazarbachi, A. Feasibility of the Combination of Venetoclax and Asparaginase-Based Chemotherapy for Adult Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2018, 18, e441–e444. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Jabbour, E.; Short, N.J.; Rausch, C.R.; Savoy, J.M.; Bose, P.; Yilmaz, M.; Jain, N.; Borthakur, G.; Ohanian, M.; et al. Clinical Experience With Venetoclax Combined With Chemotherapy for Relapsed or Refractory T-Cell Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 20, 212–218. [Google Scholar] [CrossRef]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef]
- Fogli, S.; Galimberti, S.; Gori, V.; del Re, M.; Danesi, R. Pharmacology Differences among Proteasome Inhibitors: Implications for Their Use in Clinical Practice. Pharmacol. Res. 2021, 167, 105537. [Google Scholar] [CrossRef]
- Zhang, X.; Adwal, A.; Turner, A.G.; Callen, D.F.; Abell, A.D. New Peptidomimetic Boronates for Selective Inhibition of the Chymotrypsin-like Activity of the 26S Proteasome. ACS Med. Chem. Lett. 2016, 7, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Niewerth, D.; Kaspers, G.J.L.; Jansen, G.; van Meerloo, J.; Zweegman, S.; Jenkins, G.; Whitlock, J.A.; Hunger, S.P.; Lu, X.; Alonzo, T.A.; et al. Proteasome Subunit Expression Analysis and Chemosensitivity in Relapsed Paediatric Acute Leukaemia Patients Receiving Bortezomib-Containing Chemotherapy. J. Hematol. Oncol. 2016, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Robak, P.; Robak, T. Bortezomib for the Treatment of Hematologic Malignancies: 15 Years Later. Drugs RD 2019, 19, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junk, S.; Cario, G.; Wittner, N.; Stanulla, M.; Scherer, R.; Schlegelberger, B.; Schrappe, M.; von Neuhoff, N.; Lauten, M. Bortezomib Treatment Can Overcome Glucocorticoid Resistance in Childhood B-Cell Precursor Acute Lymphoblastic Leukemia Cell Lines. Klin. Padiatr. 2015, 227, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Parker, R.; Zhang, Y.; Hawkins, E.; Kmieciak, M.; Craun, W.; Grant, S. Homoharringtonine Interacts Synergistically with Bortezomib in NHL Cells through MCL-1 and NOXA-Dependent Mechanisms. BMC Cancer 2018, 18, 1129. [Google Scholar] [CrossRef]
- Rizzatti, E.G.; Mora-Jensen, H.; Weniger, M.A.; Gibellini, F.; Lee, E.; Daibata, M.; Lai, R.; Wiestner, A. Noxa Mediates Bortezomib Induced Apoptosis in Both Sensitive and Intrinsically Resistant Mantle Cell Lymphoma Cells and This Effect Is Independent of Constitutive Activity of the AKT and NF-KappaB Pathways. Leuk. Lymphoma 2008, 49, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Ohshima-Hosoyama, S.; Davare, M.A.; Hosoyama, T.; Nelon, L.D.; Keller, C. Bortezomib Stabilizes NOXA and Triggers ROS-Associated Apoptosis in Medulloblastoma. J. Neurooncol. 2011, 105, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Z.; Ziffra, J.; Stennett, L.; Bodner, B.; Bonish, B.K.; Chaturvedi, V.; Bennett, F.; Pollock, P.M.; Trent, J.M.; Hendrix, M.J.C.; et al. Proteasome Inhibitors Trigger NOXA-Mediated Apoptosis in Melanoma and Myeloma Cells. Cancer Res. 2005, 65, 6282–6293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, M.S.; Mohammadi, M.H.; Roudsari, R.V.; Jafari, L.; Mashati, P.; Gharehbaghian, A. Proteasome Inhibition by Carfilzomib Induced Apotosis and Autophagy in a T-Cell Acute Lymphoblastic Leukemia Cell Line. Iran. J. Pharm. Res. 2019, 18, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Chandra, D.; Rudd, M.D.; Butler, A.P.; Pallotta, V.; Brown, D.; Coffer, P.J.; Tang, D.G. Induction of Prosurvival Molecules by Apoptotic Stimuli: Involvement of FOXO3a and ROS. Oncogene 2005, 24, 2020–2031. [Google Scholar] [CrossRef] [Green Version]
- Koga, T.; Suico, M.A.; Shimasaki, S.; Watanabe, E.; Kai, Y.; Koyama, K.; Omachi, K.; Morino-Koga, S.; Sato, T.; Shuto, T.; et al. Endoplasmic Reticulum (ER) Stress Induces Sirtuin 1 (SIRT1) Expression via the PI3K-Akt-GSK3β Signaling Pathway and Promotes Hepatocellular Injury. J. Biol. Chem. 2015, 290, 30366–30374. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bai, C.; Lu, D.; Wu, X.; Gao, L.; Zhang, W. Endoplasmic Reticulum Stress and Autophagy Participate in Apoptosis Induced by Bortezomib in Cervical Cancer Cells. Biotechnol. Lett. 2016, 38, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Nalluri, S.; Fiskus, W.; Savoie, A.; Buckley, K.M.; Ha, K.; Balusu, R.; Joshi, A.; Coothankandaswamy, V.; Tao, J.; et al. Role of CAAT/Enhancer Binding Protein Homologous Protein in Panobinostat-Mediated Potentiation of Bortezomib-Induced Lethal Endoplasmic Reticulum Stress in Mantle Cell Lymphoma Cells. Clin. Cancer Res. 2010, 16, 4742–4754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liang, M.; Jiang, J.; He, R.; Wang, M.; Guo, X.; Shen, M.; Qin, R. Combined Inhibition of Autophagy and Nrf2 Signaling Augments Bortezomib-Induced Apoptosis by Increasing ROS Production and ER Stress in Pancreatic Cancer Cells. Int. J. Biol. 2018, 14, 1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, M.J.; Ziegler, D.S.; Bautista Sirvent, F.J.; Attarbaschi, A.; Gore, L.; Locatelli, F.; O’Brien, M.M.; Pauly, M.; Obreja, M.; Morris, C.L.; et al. Phase 1b Study of Carfilzomib in Combination with Induction Chemotherapy in Children with Relapsed or Refractory Acute Lymphoblastic Leukemia (ALL). Blood 2019, 134, 3873. [Google Scholar] [CrossRef]
- Jonas, B.A.; Fisch, S.C.; Rosenberg, A.S.; Hoeg, R.T.; Tuscano, J.M.; Abedi, M. Phase I Study of Escalating Doses of Carfilzomib with HyperCVAD in Patients with Newly Diagnosed Acute Lymphoblastic Leukemia. Am. J. Hematol. 2021, 96, E114–E117. [Google Scholar] [CrossRef]
- Roeten, M.S.F.; van Meerloo, J.; Kwidama, Z.J.; ter Huizen, G.; Segerink, W.H.; Zweegman, S.; Kaspers, G.J.L.; Jansen, G.; Cloos, J. Pre-Clinical Evaluation of the Proteasome Inhibitor Ixazomib against Bortezomib-Resistant Leukemia Cells and Primary Acute Leukemia Cells. Cells 2021, 10, 665. [Google Scholar] [CrossRef]
- Messinger, Y.; Gaynon, P.; Raetz, E.; Hutchinson, R.; DuBois, S.; Glade-Bender, J.; Sposto, R.; van der Giessen, J.; Eckroth, E.; Bostrom, B.C. Phase I Study of Bortezomib Combined with Chemotherapy in Children with Relapsed Childhood Acute Lymphoblastic Leukemia (ALL): A Report from the Therapeutic Advances in Childhood Leukemia (TACL) Consortium. Pediatr. Blood Cancer 2010, 55, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Messinger, Y.H.; Gaynon, P.S.; Sposto, R.; van der Giessen, J.; Eckroth, E.; Malvar, J.; Bostrom, B.C. Bortezomib with Chemotherapy Is Highly Active in Advanced B-Precursor Acute Lymphoblastic Leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood 2012, 120, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Bertaina, A.; Vinti, L.; Strocchio, L.; Gaspari, S.; Caruso, R.; Algeri, M.; Coletti, V.; Gurnari, C.; Romano, M.; Cefalo, M.G.; et al. The Combination of Bortezomib with Chemotherapy to Treat Relapsed/Refractory Acute Lymphoblastic Leukaemia of Childhood. Br. J. Haematol. 2017, 176, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Horton, T.M.; Whitlock, J.A.; Lu, X.; O’Brien, M.M.; Borowitz, M.J.; Devidas, M.; Raetz, E.A.; Brown, P.A.; Carroll, W.L.; Hunger, S.P. Bortezomib Reinduction Chemotherapy in High-Risk ALL in First Relapse: A Report from the Children’s Oncology Group. Br. J. Haematol. 2019, 186, 274–285. [Google Scholar] [CrossRef]
- Burke, M.J.; Ziegler, D.S.; Bautista, F.; Attarbaschi, A.; Gore, L.; Locatelli, F.; O’Brien, M.M.; Pauly, M.; Obreja, M.; Morris, C.L.; et al. Phase 1b Study of Carfilzomib in Combination with Induction Chemotherapy in Children with Relapsed or Refractory Acute Lymphoblastic Leukemia (ALL). Blood 2021, 138, 1235. [Google Scholar] [CrossRef]
- Townsend, M.L.; Pound, M.W.; Drew, R.H. Tigecycline: A New Glycylcycline Antimicrobial. Int. J. Clin. Pract. 2006, 60, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Abbas, M.N.; Kausar, S.; Yang, J.; Li, L.; Tan, L.; Cui, H. Biological Functions and Molecular Mechanisms of Antibiotic Tigecycline in the Treatment of Cancers. Int. J. Mol. Sci. 2019, 20, 3577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting Mitochondrial Oxidative Phosphorylation Eradicates-Resistant Chronic Myeloid Leukemic Stem Cells. Nat. Med. 2017, 23, 1234. [Google Scholar] [CrossRef] [Green Version]
- Samuels, A.L.; Heng, J.Y.; Beesley, A.H.; Kees, U.R. Bioenergetic Modulation Overcomes Glucocorticoid Resistance in T-Lineage Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2014, 165, 57–66. [Google Scholar] [CrossRef]
- Fu, X.; Liu, W.; Huang, Q.; Wang, Y.; Li, H.; Xiong, Y. Targeting Mitochondrial Respiration Selectively Sensitizes Pediatric Acute Lymphoblastic Leukemia Cell Lines and Patient Samples to Standard Chemotherapy. Am. J. Cancer Res. 2017, 7, 2395. [Google Scholar]
- Bogush, T.A.; Polezhaev, B.B.; Mamichev, I.A.; Bogush, E.A.; Polotsky, B.E.; Tjulandin, S.A.; Ryabov, A.B. Tamoxifen Never Ceases to Amaze: New Findings on Non-Estrogen Receptor Molecular Targets and Mediated Effects. Cancer Investig. 2018, 36, 211–220. [Google Scholar] [CrossRef]
- Bogush, T.; Dudko, E.; Bogush, E.; Polotsky, B.; Tjulandin, S.; Davydov, M. Tamoxifen Non-Estrogen Receptor Mediated Molecular Targets. Oncol. Rev. 2012, 6, 122–129. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.J.; Gorski, S.M. Molecular Mechanisms Underlying Autophagy-Mediated Treatment Resistance in Cancer. Cancers 2019, 11, 1775. [Google Scholar] [CrossRef] [Green Version]
- Torres-López, L.; Maycotte, P.; Liñán-Rico, A.; Liñán-Rico, L.; Donis-Maturano, L.; Delgado-Enciso, I.; Meza-Robles, C.; Vásquez-Jiménez, C.; Hernández-Cruz, A.; Dobrovinskaya, O. Tamoxifen Induces Toxicity, Causes Autophagy, and Partially Reverses Dexamethasone Resistance in Jurkat T Cells. J. Leukoc. Biol. 2019, 105, 983–998. [Google Scholar] [CrossRef]
- Szaflarski, J.P.; Bebin, E.M.; Comi, A.M.; Patel, A.D.; Joshi, C.; Checketts, D.; Beal, J.C.; Laux, L.C.; de Boer, L.M.; Wong, M.H.; et al. Long-term Safety and Treatment Effects of Cannabidiol in Children and Adults with Treatment-resistant Epilepsies: Expanded Access Program Results. Epilepsia 2018, 59, 1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massi, P.; Solinas, M.; Cinquina, V.; Parolaro, D. Cannabidiol as Potential Anticancer Drug. Br. J. Clin. Pharmacol. 2013, 75, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimmerman, N.; Ben-Hail, D.; Porat, Z.; Juknat, A.; Kozela, E.; Daniels, M.P.; Connelly, P.S.; Leishman, E.; Bradshaw, H.B.; Shoshan-Barmatz, V.; et al. Direct Modulation of the Outer Mitochondrial Membrane Channel, Voltage-Dependent Anion Channel 1 (VDAC1) by Cannabidiol: A Novel Mechanism for Cannabinoid-Induced Cell Death. Cell Death Dis. 2013, 4, e949. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M. VDAC in Cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 665–673. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Pottosin, I.; Dobrovinskaya, O. Mitochondria as Emerging Targets for Therapies against T Cell Acute Lymphoblastic Leukemia. J. Leukoc. Biol. 2019, 105, 935–946. [Google Scholar] [CrossRef]
- Wang, X.; Lou, K.; Song, X.; Ma, H.; Zhou, X.; Xu, H.; Wang, W. Mebendazole Is a Potent Inhibitor to Chemoresistant T Cell Acute Lymphoblastic Leukemia Cells. Toxicol. Appl. Pharmacol. 2020, 396, 115001. [Google Scholar] [CrossRef]
- Real, P.J.; Tosello, V.; Palomero, T.; Castillo, M.; Hernando, E.; de Stanchina, E.; Sulis, M.L.; Barnes, K.; Sawai, C.; Homminga, I.; et al. γ-Secretase Inhibitors Reverse Glucocorticoid Resistance in T Cell Acute Lymphoblastic Leukemia. Nat. Med. 2008, 15, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Jing, D.; Huang, Y.; Liu, X.; Sia, K.C.S.; Zhang, J.C.; Tai, X.; Wang, M.; Toscan, C.E.; McCalmont, H.; Evans, K.; et al. Lymphocyte-Specific Chromatin Accessibility Pre-Determines Glucocorticoid Resistance in Acute Lymphoblastic Leukemia. Cancer Cell 2018, 34, 906–921.e8. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Drugs | Mechanism of Action | Preclinical Studies | Completed Clinical Studies | Ongoing Clinical Studies |
|---|---|---|---|---|
| MK2206 | Allosteric AKT 1 inhibition | MK2206 and dexamethasone co-treatment of resistant ALL 2 cell lines in vitro and in vivo [99] | Not applicable | Not applicable |
| Pictilisib Idelalisib Buparlisib ZSTK-474 AS-605240 CAL-101 Duvelisib | pan-PI3K 3 inhibition | Pictilisib in T-ALL 4 treatment [102,103,104,105] Idelalisib treatment of T-cell leukemia-lymphoma samples [107] | Not applicable | Phase I, chimeric antigen receptor T-cell followed by duvelisib for ALL, LL 5, and lymphosarcoma (NCT05044039). Phase I, duvelisib for relapsing/remitting T-LL 7 |
| Buparlisib treatment of T-ALL cell lines [108,109] ZSTK-474, AS-605240, CAL-101, and IPI-145 for the treatment of pre-B ALL 8 cell lines and ex vivo blasts from pre-B ALL patients [110] | ||||
| Dactolisib | pan-PI3K inhibition, mTOR 9 inhibition | Dactolisib treatment of T-ALL and pre-B ALL cell lines [132] | Phase I, relapsed/remitting ALL [115] | Not applicable |
| Ruxolitinib | JAK1/2 inhibition | Ruxolitinib and dexamethasone co-treatment of T-ALL cells [90] Ruxolitinib and VXL 10 chemotherapy in mice engrafted with Ph-like 11 ALL [96] Ruxolitinib treatment of cell lines with IL-7 12-signaling, mediated steroid resistance. Ruxolitinib alone or in co-treatment with prednisolone in the T-ALL xenografts ex vivo [118] Venetoclax and ruxolitinib co-treatment in vivo in treating mice engrafted with blasts carrying JAK3 mutation [131] | Phase II—part 1, ruxolitinib and consolidation chemotherapy for pediatric Ph-like ALL [133] | Phase I, newly diagnosed pediatric Ph-like ALL (NCT03571321) Phase II—part 2, pediatric Ph-like ALL (NCT02723994) Phase II, relapsing/remitting Ph-like ALL (NCT02420717) Phase II/III, pediatric T-ALL/T-LL or pre-B ALL/B-LL 13 (NCT03117751) |
| CI1040 | MAPK 14 inhibition | CI1040 treatment of glucocorticoid-resistant cell lines [89] CI1040 treatment of cell lines with IL-7-signaling, mediated steroid resistance [118] | Not applicable | Not applicable |
| Selumetinib Trametinib Binimetinib | MAPK inhibition | Selumetinib and trametinib treatment of cell lines with IL-7-signaling, mediated steroid resistance. Selumetinib, trametinib and binimetinib alone in the T-ALL xenografts ex vivo. Selumetinib in co-treatment with prednisolone alone in the T-ALL xenografts ex vivo [118] Selumetinib ex vivo treatment of pre-B ALL samples. Selumetinib in vivo treatment of xenografts with Ras 15 pathway mutant/wild-type ALL cells [120] Selumetinib in vivo and in vitro treatment of xenografts with Ras pathway mutant ALL cells [122] Selumetinib in vivo and in vitro treatment of xenografts with Ras pathway mutant/wild-type ALL cells [122] Selumetinib in co-treatment with dexamethasone for pre-B ALL samples ex vivo and in vitro pre-B ALL and T-ALL cell lines [123] Trametinib treatment ex vivo for pre-B ALL samples [121] Selumetinib, trametinib and binimetinib treatment of Ras mutated/wild-type MLL 16 rearranged ALL cell lines [128] Tofacitinib and MAPK inhibitors co-treatment for JAK 6-mutated ALL cell lines and ex vivo blasts from T-ALL patients [131] | Not applicable | Phase I/II, selumetinib for the relapsing/remitting ALL (NCT03705507) |
| Drug | Preclinical Studies | Completed Clinical Studies | Ongoing Clinical Studies |
|---|---|---|---|
| Venetoclax | Venetoclax treatment of T-ALL 1 and ETP ALL 2 cell lines/blasts from patients [143] Venetoclax treatment of T-ALL cell lines/blasts from patients [144] | Series of cases, relapsing/remitting ALL 3 [145] Retrospective study, relapsing/remitting T-ALL [146] Phase I, relapsing/remitting ALL [147] | Phase Ib-II, navitoclax and venetoclax co-treatment for pre-transplant and post-transplant treatment of adult T-ALL patients (NCT05054465) Phase I, adult pre-B ALL 4 (NCT05157971) Phase I/phase II, relapsing/remitting ALL (NCT03808610, NCT03504644, NCT03576547, NCT03319901, NCT04872790, NCT05016947, NCT03808610, NCT04752163, and NCT05149378) |
| Venetoclax and tofacitinib ex vivo co-treatment of JAK 5-mutated ALL cell lines and blasts from T-ALL patients [137] | |||
| Navitoclax | Navitoclax treatment of T-ALL and ETP ALL cell lines/blasts from patients [143] | Phase I, relapsing/remitting ALL [147] | Phase Ib-II, navitoclax and venetoclax co-treatment for pre-transplant and post-transplant treatment of adult T-ALL patients (NCT05054465) |
| Drug | Preclinical Studies | Completed Clinical Studies | Ongoing Clinical Studies |
| Bortezomib | Bortezomib and prednisone co-treatment of resistant/sensitive ALL 1 cell lines [152] Bortezomib treatment of sensitive T-ALL cell line and cells from primary ALL patients [166] | Phase I/II, pediatric relapsed ALL [167,168] Prospective cohort study, pediatric relapsed ALL [169] Phase II, relapsed ALL/T-LL [170] | Phase II, infants with newly diagnosed ALL (NCT02553460) Phase II/III, pediatric T-ALL 2/T-LL 3 or pre-B ALL 4/B-LL 5 (NCT03117751) Phase III, newly diagnosed ALL/LL 6 (NCT02112916) AIEOP-BFM ALL 2017 study, early- HR 7 pre-B ALL (NCT03390387) Phase II, relapsed/remitting ALL (NCT03136146, NCT03590171) Phase IV, relapsed ALL (NCT05137860) |
| Carfilzomib | Carfilzomib and dexamethasone co-treatment in resistant cell line [157] | Phase I, relapsed/refractory ALL [171] | Phase II, relapsed/refractory ALL (NCT02303821) |
| Phase I, newly diagnosed ALL [165] | Phase I, relapsed/refractory solid tumors or leukemia (NCT02512926) | ||
| Ixazomib | Ixazomib treatment of sensitive T-ALL cell line and cells from primary ALL patients [166] | Not applicable | Phase I/II, relapsed/remitting ALL (NCT03817320) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kośmider, K.; Karska, K.; Kozakiewicz, A.; Lejman, M.; Zawitkowska, J. Overcoming Steroid Resistance in Pediatric Acute Lymphoblastic Leukemia—The State-of-the-Art Knowledge and Future Prospects. Int. J. Mol. Sci. 2022, 23, 3795. https://doi.org/10.3390/ijms23073795
Kośmider K, Karska K, Kozakiewicz A, Lejman M, Zawitkowska J. Overcoming Steroid Resistance in Pediatric Acute Lymphoblastic Leukemia—The State-of-the-Art Knowledge and Future Prospects. International Journal of Molecular Sciences. 2022; 23(7):3795. https://doi.org/10.3390/ijms23073795
Chicago/Turabian StyleKośmider, Kamil, Katarzyna Karska, Agata Kozakiewicz, Monika Lejman, and Joanna Zawitkowska. 2022. "Overcoming Steroid Resistance in Pediatric Acute Lymphoblastic Leukemia—The State-of-the-Art Knowledge and Future Prospects" International Journal of Molecular Sciences 23, no. 7: 3795. https://doi.org/10.3390/ijms23073795