
Metabolic Alterations in Cellular Senescence: The Role of Citrate in Ageing and Age-Related Disease


{kind=link}
Abstract
:1. Introduction
1.1. Cellular Senescence
1.2. The Senescence—Associated Secretory Phenotype (SASP)
1.3. Telomeres and Senescence
1.4. Telomere Dysfunction Regulates Mitochondrial Function and ROS and Vice Versa
1.5. Dietary Factors and the Regulation of Nicotinamide Adenine Dinucleotide (NAD+), Telomere and Mitochondrial Dysfunction and Its Relationship to Senescence
2. Senescent Cell Metabolism
2.1. Glycolysis and Pyruvate
2.2. Energy Regulation in Oncogene-Induced Senescence
2.3. Energy Regulation in Replicative Senescence
2.4. Unbiased Metabolomic Screens of Senescent Cells
2.5. Do Senescent Cells Exhibit a Reverse TCA Cycle?
2.6. The Relationship of the Extracellular Senescence Metabolome to the Internal Senescence Metabolome
3. The Role of p53 in Fibroblast Senescence and Metabolism
P53 Decline in Classical Genotoxic SIPS May Be Associated with a Shift towards Glycolysis and the PPP
4. The Extracellular Senescence Metabolome and the Identification of Extracellular Citrate Accumulation as a Common Feature of Replicative Senescence and Senescence-Induced by Irreparable DNA Damage: Evidence for Cell Type Specificity
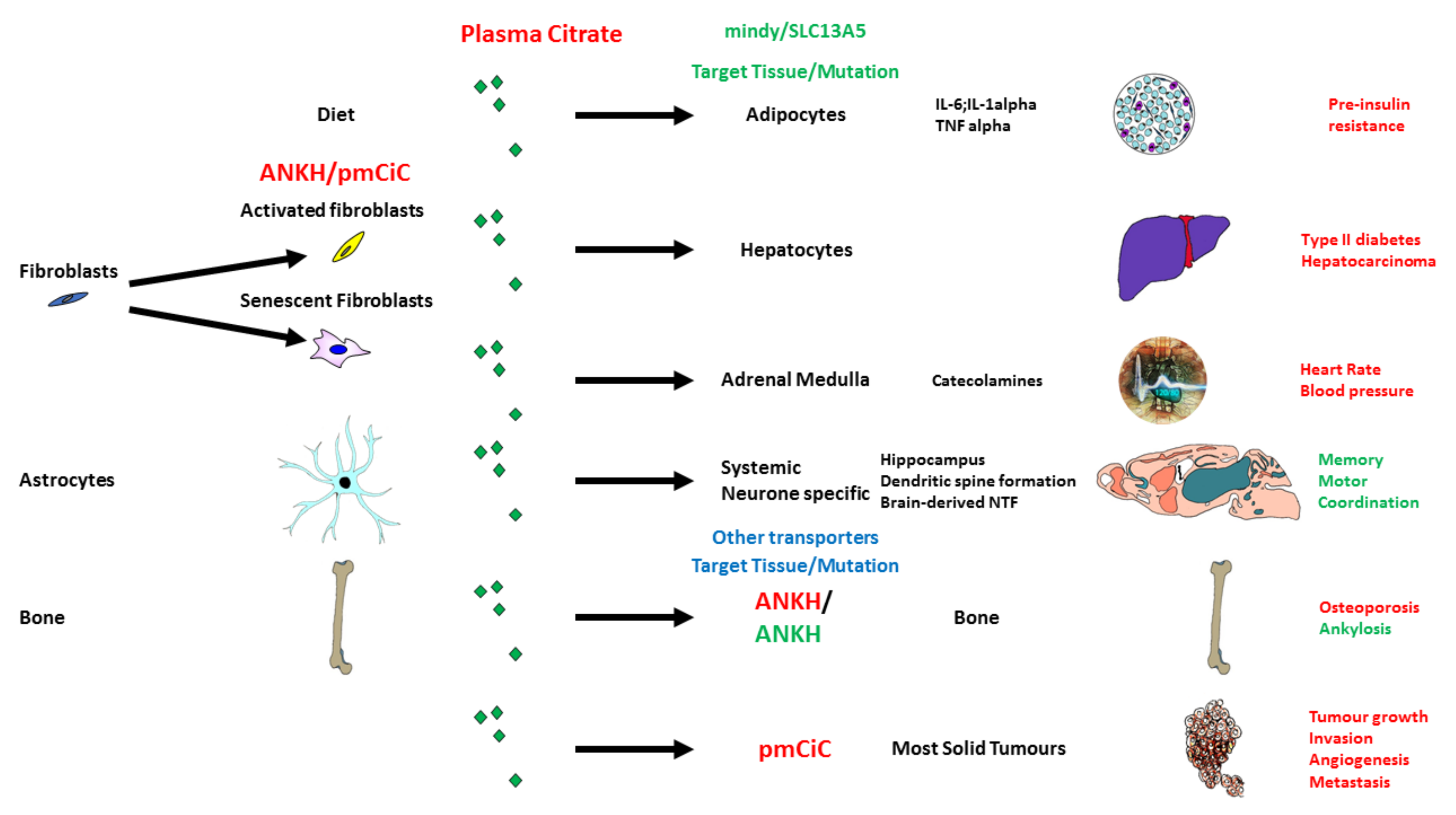
5. Citrate Transport and Its Role in Ageing and Disease
5.1. Citrate in the Diet and Its Potential Effect on Senescence and the SASP
5.2. Citrate Import by SLC13A5/I’m Not Dead Yet (INDY)
5.3. Citrate Export and Its Upregulation in Senescence and Fibroblast Activation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Sherr, C.J.; DePinho, R.A. Cellular senescence: Mitotic clock or culture shock? Cell 2000, 102, 407–410. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl. Acad. Sci. USA 2016, 113, E5024–E5033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Lowe, R.; Overhoff, M.G.; Ramagopalan, S.V.; Garbe, J.C.; Koh, J.; Stampfer, M.R.; Beach, D.H.; Rakyan, V.K.; Bishop, C.L. The senescent methylome and its relationship with cancer, ageing and germline genetic variation in humans. Genome Biol. 2015, 16, 194. [Google Scholar] [CrossRef] [Green Version]
- Tyler, E.J.; Gutierrez Del Arroyo, A.; Hughes, B.K.; Wallis, R.; Garbe, J.C.; Stampfer, M.R.; Koh, J.; Lowe, R.; Philpott, M.P.; Bishop, C.L. Early growth response 2 (EGR2) is a novel regulator of the senescence programme. Aging Cell 2021, 20, e13318. [Google Scholar] [CrossRef]
- Munro, J.; Steeghs, K.; Morrison, V.; Ireland, H.; Parkinson, E.K. Human fibroblast replicative senescence can occur in the absence of extensive cell division and short telomeres. Oncogene 2001, 20, 3541–3552. [Google Scholar] [CrossRef] [Green Version]
- Marthandan, S.; Priebe, S.; Hemmerich, P.; Klement, K.; Diekmann, S. Long-term quiescent fibroblast cells transit into senescence. PLoS ONE 2014, 9, e115597. [Google Scholar] [CrossRef] [Green Version]
- Jacome Burbano, M.S.; Gilson, E. Long-lived post-mitotic cell aging: Is a telomere clock at play? Mech. Ageing Dev. 2020, 189, 111256. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Schafer, M.J.; Noren Hooten, N.; Atkinson, E.J.; Evans, M.K.; Baker, D.J.; Quarles, E.K.; Robbins, P.D.; Ladiges, W.C.; LeBrasseur, N.K.; et al. Circulating levels of monocyte chemoattractant protein-1 as a potential measure of biological age in mice and frailty in humans. Aging Cell 2018, 17, e12706. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Kratz, K.; de Lange, T. Protection of telomeres 1 proteins POT1a and POT1b can repress ATR signaling by RPA exclusion, but binding to CST limits ATR repression by POT1b. J. Biol. Chem. 2018, 293, 14384–14392. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Allsopp, R.C.; Chang, E.; Kashefi-Aazam, M.; Rogaev, E.I.; Piatyszek, M.A.; Shay, J.W.; Harley, C.B. Telomere shortening is associated with cell division in vitro and in vivo. Exp. Cell Res. 1995, 220, 194–200. [Google Scholar] [CrossRef]
- Kaul, Z.; Cesare, A.J.; Huschtscha, L.I.; Neumann, A.A.; Reddel, R.R. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2011, 13, 52–59. [Google Scholar] [CrossRef] [PubMed]
- D’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Aeby, E.; Ahmed, W.; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 protects telomeres from oxidative damage and preserves telomeric DNA for extension by telomerase. Cell Rep. 2016, 17, 3107–3114. [Google Scholar] [CrossRef] [Green Version]
- Kharbanda, S.; Kumar, V.; Dhar, S.; Pandey, P.; Chen, C.; Majumder, P.; Yuan, Z.M.; Whang, Y.; Strauss, W.; Pandita, T.K.; et al. Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr. Biol. 2000, 10, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012, 31, 2839–2851. [Google Scholar] [CrossRef] [PubMed]
- Bayne, S.; Liu, J.P. Hormones and growth factors regulate telomerase activity in ageing and cancer. Mol. Cell Endocrinol. 2005, 240, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Epel, E.S.; Blackburn, E.H.; Lin, J.; Dhabhar, F.S.; Adler, N.E.; Morrow, J.D.; Cawthon, R.M. Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. USA 2004, 101, 17312–17315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhan, Y.; Pedersen, N.L.; Fang, F.; Hagg, S. Telomere length and all-cause mortality: A meta-analysis. Ageing Res. Rev. 2018, 48, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Denham, J.; McCluskey, M.; Denham, M.M.; Sellami, M.; Davie, A.J. Epigenetic control of exercise adaptations in the equine athlete: Current evidence and future directions. Equine Vet. J. 2021, 53, 431–450. [Google Scholar] [CrossRef]
- Sellami, M.; Bragazzi, N.; Prince, M.S.; Denham, J.; Elrayess, M. Regular, intense exercise training as a healthy aging lifestyle strategy: Preventing DNA damage, telomere shortening and adverse DNA methylation changes over a lifetime. Front. Genet. 2021, 12, 652497. [Google Scholar] [CrossRef]
- Tsoukalas, D.; Fragkiadaki, P.; Docea, A.O.; Alegakis, A.K.; Sarandi, E.; Thanasoula, M.; Spandidos, D.A.; Tsatsakis, A.; Razgonova, M.P.; Calina, D. Discovery of potent telomerase activators: Unfolding new therapeutic and anti-aging perspectives. Mol. Med. Rep. 2019, 20, 3701–3708. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Rudolph, K.L.; Chang, S.; Lee, H.W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Kipling, D.; Faragher, R.G. Telomeres. Ageing hard or hardly ageing? Nature 1999, 398, 191–193. [Google Scholar] [CrossRef]
- Dokal, I. Dyskeratosis congenita. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.Y.; Armanios, M.; Strong, M.A.; Karim, B.; Feldser, D.M.; Huso, D.; Greider, C.W. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 2005, 123, 1121–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadalla, S.M.; Cawthon, R.; Giri, N.; Alter, B.P.; Savage, S.A. Telomere length in blood, buccal cells, and fibroblasts from patients with inherited bone marrow failure syndromes. Aging 2010, 2, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.R.; Wood, E.; Collins, K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 1999, 402, 551–555. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Lorente, M.A.; Cano-Martin, A.C.; Blasco, M.A. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 2019, 10, 4723. [Google Scholar] [CrossRef] [Green Version]
- Veatch, J.R.; McMurray, M.A.; Nelson, Z.W.; Gottschling, D.E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 2009, 137, 1247–1258. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Saretzki, G.; von Zglinicki, T. DNA damage in telomeres and mitochondria during cellular senescence: Is there a connection? Nucleic Acids Res. 2007, 35, 7505–7513. [Google Scholar] [CrossRef]
- Passos, J.F.; von Zglinicki, T.; Saretzki, G. Mitochondrial dysfunction and cell senescence: Cause or consequence? Rejuvenation Res. 2006, 9, 64–68. [Google Scholar] [CrossRef]
- Velarde, M.C.; Demaria, M.; Melov, S.; Campisi, J. Pleiotropic age-dependent effects of mitochondrial dysfunction on epidermal stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 10407–10412. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef] [PubMed]
- Harle-Bachor, C.; Boukamp, P. Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proc. Natl. Acad. Sci. USA 1996, 93, 6476–6481. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, R.D.; Wright, W.E.; Shay, J.W.; Taylor, R.S. Telomerase activity concentrates in the mitotically active segments of human hair follicles. J. Investig. Dermatol. 1997, 108, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Dellambra, E.; Golisano, O.; Bondanza, S.; Siviero, E.; Lacal, P.; Molinari, M.; D’Atri, S.; De Luca, M. Downregulation of 14-3-3sigma prevents clonal evolution and leads to immortalization of primary human keratinocytes. J. Cell Biol. 2000, 149, 1117–1130. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.C.; Ji, X.; Nissim, I.; Long, F. Malic enzyme couples mitochondria with aerobic glycolysis in osteoblasts. Cell Rep. 2020, 32, 108108. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Imai, S.I. NAD (+) biosynthesis, aging, and disease. F1000Research 2018, 7, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quijano, C.; Cao, L.; Fergusson, M.M.; Romero, H.; Liu, J.; Gutkind, S.; Rovira, I.I.; Mohney, R.P.; Karoly, E.D.; Finkel, T. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 2012, 11, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- James, E.L.; Lane, J.A.; Michalek, R.D.; Karoly, E.D.; Parkinson, E.K. Replicatively senescent human fibroblasts reveal a distinct intracellular metabolic profile with alterations in NAD+ and nicotinamide metabolism. Sci. Rep. 2016, 6, 38489. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Wang, K.; Stock, A.J.; Gong, Y.; Demarest, T.G.; Yang, B.; Giri, N.; Harrington, L.; Alter, B.P.; Savage, S.A.; et al. Re-equilibration of imbalanced NAD metabolism ameliorates the impact of telomere dysfunction. EMBO J. 2020, 39, e103420. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Yang, B.; Dan, X.; Hou, Y.; Lee, J.H.; Wechter, N.; Krishnamurthy, S.; Kimura, R.; Babbar, M.; Demarest, T.; McDevitt, R.; et al. NAD(+) supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 2021, 20, e13329. [Google Scholar] [CrossRef]
- Clement, J.; Wong, M.; Poljak, A.; Sachdev, P.; Braidy, N. The plasma NAD(+) metabolome is dysregulated in “normal” aging. Rejuvenation Res. 2019, 22, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhassan, Y.S.; Kluckova, K.; Fletcher, R.S.; Schmidt, M.S.; Garten, A.; Doig, C.L.; Cartwright, D.M.; Oakey, L.; Burley, C.V.; Jenkinson, N.; et al. Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 2019, 28, 1717–1728.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapata-Perez, R.; Wanders, R.J.A.; van Karnebeek, C.D.M.; Houtkooper, R.H. NAD(+) homeostasis in human health and disease. EMBO Mol. Med. 2021, 13, e13943. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, S.; Drexler, K.; Federlin, M.; Schlitt, H.J.; Berneburg, M.; Adamski, J.; Gaumann, A.; Geissler, E.K.; Ganapathy, V.; Parkinson, E.K.; et al. Extracellular citrate fuels cancer cell metabolism and growth. Front. Cell Dev. Biol 2020, 8, 602476. [Google Scholar] [CrossRef]
- Zwerschke, W.; Mazurek, S.; Stockl, P.; Hutter, E.; Eigenbrodt, E.; Jansen-Durr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 2003, 376, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, E.K.; Adamski, J.; Zahn, G.; Gaumann, A.; Flores-Borja, F.; Ziegler, C.; Mycielska, M.E. Extracellular citrate and metabolic adaptations of cancer cells. Cancer Metastasis. Rev. 2021, 40, 1073–1091. [Google Scholar] [CrossRef]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Nakashima, Y.; Yokode, M.; Tanaka, M.; Bernard, D.; Gil, J.; Beach, D. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid. Redox Signal. 2007, 9, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2014, 19, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Guo, Y.; Fan, Y.; Wang, Q.; Zhang, Q.; Lai, D. Decreased expression of IDH1 by chronic unpredictable stress suppresses proliferation and accelerates senescence of granulosa cells through ROS activated MAPK signaling pathways. Free Radic. Biol. Med. 2021, 169, 122–136. [Google Scholar] [CrossRef]
- Wiley, C.D.; Sharma, R.; Davis, S.S.; Lopez-Dominguez, J.A.; Mitchell, K.P.; Wiley, S.; Alimirah, F.; Kim, D.E.; Payne, T.; Rosko, A.; et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell. Metab. 2021, 33, 1124–1136.e5. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell. Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Robles, S.J.; Adami, G.R. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998, 16, 1113–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell. 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auro, K.; Joensuu, A.; Fischer, K.; Kettunen, J.; Salo, P.; Mattsson, H.; Niironen, M.; Kaprio, J.; Eriksson, J.G.; Lehtimaki, T.; et al. A metabolic view on menopause and ageing. Nat. Commun. 2014, 5, 4708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menni, C.; Kastenmuller, G.; Petersen, A.K.; Bell, J.T.; Psatha, M.; Tsai, P.C.; Gieger, C.; Schulz, H.; Erte, I.; John, S.; et al. Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int. J. Epidemiol. 2013, 42, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, E.; Vousden, K.H. p53 regulation of metabolic pathways. Cold Spring Harb. Perspect. Biol. 2010, 2, a001040. [Google Scholar] [CrossRef] [Green Version]
- Minakata, S.; Inai, Y.; Manabe, S.; Nishitsuji, K.; Ito, Y.; Ihara, Y. Monomeric C-mannosyl tryptophan is a degradation product of autophagy in cultured cells. Glycoconj. J. 2020, 37, 635–645. [Google Scholar] [CrossRef]
- Sakurai, S.; Inai, Y.; Minakata, S.; Manabe, S.; Ito, Y.; Ihara, Y. A novel assay for detection and quantification of C-mannosyl tryptophan in normal or diabetic mice. Sci. Rep. 2019, 9, 4675. [Google Scholar] [CrossRef]
- Hocher, B.; Adamski, J. Metabolomics for clinical use and research in chronic kidney disease. Nat. Rev. Nephrol. 2017, 13, 269–284. [Google Scholar] [CrossRef]
- James, E.N.L.; Bennett, M.H.; Parkinson, E.K. The induction of the fibroblast extracellular senescence metabolome is a dynamic process. Sci. Rep. 2018, 8, 12148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Delfarah, A.; Parrish, S.; Junge, J.A.; Yang, J.; Seo, F.; Li, S.; Mac, J.; Wang, P.; Fraser, S.E.; Graham, N.A. Inhibition of nucleotide synthesis promotes replicative senescence of human mammary epithelial cells. J. Biol. Chem. 2019, 294, 10564–10578. [Google Scholar] [CrossRef] [PubMed]
- Leandro, J.G.; Espindola-Netto, J.M.; Vianna, M.C.; Gomez, L.S.; DeMaria, T.M.; Marinho-Carvalho, M.M.; Zancan, P.; Paula Neto, H.A.; Sola-Penna, M. Exogenous citrate impairs glucose tolerance and promotes visceral adipose tissue inflammation in mice. Br. J. Nutr. 2016, 115, 967–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Mitchell, S.E.; Wang, B.; Tosti, V.; van Vliet, T.; Veronese, N.; Bertozzi, B.; Early, D.S.; Maissan, P.; Speakman, J.R.; et al. The effects of graded caloric restriction: XII. Comparison of mouse to human impact on cellular senescence in the colon. Aging Cell 2018, 17, e12746. [Google Scholar] [CrossRef] [PubMed]
- Ashbrook, M.J.; McDonough, K.L.; Pituch, J.J.; Christopherson, P.L.; Cornell, T.T.; Selewski, D.T.; Shanley, T.P.; Blatt, N.B. Citrate modulates lipopolysaccharide-induced monocyte inflammatory responses. Clin. Exp. Immunol. 2015, 180, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Rogina, B.; Reenan, R.A.; Nilsen, S.P.; Helfand, S.L. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 2000, 290, 2137–2140. [Google Scholar] [CrossRef]
- Marden, J.H.; Rogina, B.; Montooth, K.L.; Helfand, S.L. Conditional tradeoffs between aging and organismal performance of Indy long-lived mutant flies. Proc. Natl. Acad. Sci. USA 2003, 100, 3369–3373. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Fei, Y.J.; Zhuang, L.; Gopal, E.; Miyauchi, S.; Ganapathy, V. Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem. J. 2004, 378, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Zhuang, L.; Ganapathy, V. Human Na+ -coupled citrate transporter: Primary structure, genomic organization, and transport function. Biochem. Biophys. Res. Commun. 2002, 299, 465–471. [Google Scholar] [CrossRef]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef] [Green Version]
- Birkenfeld, A.L.; Lee, H.Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Fischer-Rosinsky, A.; et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Willmes, D.M.; Daniels, M.; Kurzbach, A.; Lieske, S.; Bechmann, N.; Schumann, T.; Henke, C.; El-Agroudy, N.N.; Da Costa Goncalves, A.C.; Peitzsch, M.; et al. The longevity gene mIndy (I’m Not Dead, Yet) affects blood pressure through sympathoadrenal mechanisms. JCI Insight 2021, 6, 136083. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.Z.; Sung, C.W.; Tsai, Y.H.; Yeh, S.R.; Lin, W.S.; Wang, P.Y. Nervous system deletion of mammalian INDY in mice mimics dietary restriction-induced memory enhancement. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Setti, S.E.; Hunsberger, H.C.; Reed, M.N. Alterations in hippocampal activity and Alzheimer’s Disease. Transl. Issues Psychol. Sci. 2017, 3, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Huard, K.; Gosset, J.R.; Montgomery, J.I.; Gilbert, A.; Hayward, M.M.; Magee, T.V.; Cabral, S.; Uccello, D.P.; Bahnck, K.; Brown, J.; et al. Optimization of a dicarboxylic series for in vivo inhibition of citrate transport by the solute carrier 13 (SLC13) family. J. Med. Chem. 2016, 59, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Bliuc, D.; Tran, T.; Adachi, J.D.; Atkins, G.J.; Berger, C.; van den Bergh, J.; Cappai, R.; Eisman, J.A.; van Geel, T.; Geusens, P.; et al. Cognitive decline is associated with an accelerated rate of bone loss and increased fracture risk in women: A prospective study from the Canadian Multicentre Osteoporosis Study. J. Bone Miner. Res. 2021, 36, 2106–2115. [Google Scholar] [CrossRef]
- Lary, C.W.; Rosen, C.J.; Kiel, D.P. Osteoporosis and Dementia: Establishing a link. J. Bone Miner. Res. 2021, 36, 2103–2105. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Kopel, J.J.; Lawrence, J.J.; Neugebauer, V.; Ganapathy, V. Plasma membrane Na(+)-coupled citrate transporter (SLC13A5) and neonatal epileptic encephalopathy. Molecules 2017, 22, 378. [Google Scholar] [CrossRef]
- Kopel, J.J.; Bhutia, Y.D.; Sivaprakasam, S.; Ganapathy, V. Consequences of NaCT/SLC13A5/mINDY deficiency: Good versus evil, separated only by the blood-brain barrier. Biochem. J. 2021, 478, 463–486. [Google Scholar] [CrossRef]
- Hardies, K.; de Kovel, C.G.; Weckhuysen, S.; Asselbergh, B.; Geuens, T.; Deconinck, T.; Azmi, A.; May, P.; Brilstra, E.; Becker, F.; et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 2015, 138, 3238–3250. [Google Scholar] [CrossRef] [Green Version]
- Thevenon, J.; Milh, M.; Feillet, F.; St-Onge, J.; Duffourd, Y.; Juge, C.; Roubertie, A.; Heron, D.; Mignot, C.; Raffo, E.; et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 2014, 95, 113–120. [Google Scholar] [CrossRef]
- Westergaard, N.; Waagepetersen, H.S.; Belhage, B.; Schousboe, A. Citrate, a ubiquitous key metabolite with regulatory function in the cNS. Neurochem. Res. 2017, 42, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Gopal, E.; Babu, E.; Ramachandran, S.; Bhutia, Y.D.; Prasad, P.D.; Ganapathy, V. Species-specific influence of lithium on the activity of SLC13A5 (NaCT): Lithium-induced activation is specific for the transporter in primates. J. Pharmacol. Exp. Ther. 2015, 353, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espanhol, J.C.L.; Vieira-Coelho, M.A. Effects of lithium use on the white matter of patients with bipolar disorder—A systematic review. Nord. J. Psychiatry 2021, 76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- von Loeffelholz, C.; Lieske, S.; Neuschafer-Rube, F.; Willmes, D.M.; Raschzok, N.; Sauer, I.M.; Konig, J.; Fromm, M.F.; Horn, P.; Chatzigeorgiou, A.; et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 2017, 66, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, J.P. Isocitric and citric acid in human prostatic and seminal fluid: Implications for prostatic metabolism and secretion. Prostate 1994, 24, 139–142. [Google Scholar] [CrossRef]
- Mazurek, M.P.; Prasad, P.D.; Gopal, E.; Fraser, S.P.; Bolt, L.; Rizaner, N.; Palmer, C.P.; Foster, C.S.; Palmieri, F.; Ganapathy, V.; et al. Molecular origin of plasma membrane citrate transporter in human prostate epithelial cells. EMBO Rep. 2010, 11, 431–437. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Dettmer, K.; Rummele, P.; Schmidt, K.; Prehn, C.; Milenkovic, V.M.; Jagla, W.; Madej, G.M.; Lantow, M.; Schladt, M.; et al. Extracellular citrate affects critical elements of cancer cell metabolism and supports cancer development in vivo. Cancer Res. 2018, 78, 2513–2523. [Google Scholar] [CrossRef] [Green Version]
- Drexler, K.; Schmidt, K.M.; Jordan, K.; Federlin, M.; Milenkovic, V.M.; Liebisch, G.; Artati, A.; Schmidl, C.; Madej, G.; Tokarz, J.; et al. Cancer-associated cells release citrate to support tumour metastatic progression. Life Sci. Alliance 2021, 4, e202000903. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Z.; Chen, V.P.; Wang, L.; Inman, C.L.; Zhou, Y.; Guo, C.; Tchkonia, T.; Rowe, D.W.; Kuchel, G.A.; et al. Transplanting cells from old but not young donors causes physical dysfunction in older recipients. Aging Cell 2020, 19, e13106. [Google Scholar] [CrossRef] [PubMed]
- Mycielska, M.E.; Patel, A.; Rizaner, N.; Mazurek, M.P.; Keun, H.; Ganapathy, V.; Djamgoz, M.B. Citrate transport and metabolism in mammalian cells: Prostate epithelial cells and prostate cancer. Bioessays 2009, 31, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Castro, P.; Giri, D.; Lamb, D.; Ittmann, M. Cellular senescence in the pathogenesis of benign prostatic hyperplasia. Prostate 2003, 55, 30–38. [Google Scholar] [CrossRef]
- Choi, J.; Shendrik, I.; Peacocke, M.; Peehl, D.; Buttyan, R.; Ikeguchi, E.F.; Katz, A.E.; Benson, M.C. Expression of senescence-associated beta-galactosidase in enlarged prostates from men with benign prostatic hyperplasia. Urology 2000, 56, 160–166. [Google Scholar] [CrossRef]
- Untergasser, G.; Gander, R.; Rumpold, H.; Heinrich, E.; Plas, E.; Berger, P. TGF-beta cytokines increase senescence-associated beta-galactosidase activity in human prostate basal cells by supporting differentiation processes, but not cellular senescence. Exp. Gerontol. 2003, 38, 1179–1188. [Google Scholar] [CrossRef]
- Szeri, F.; Lundkvist, S.; Donnelly, S.; Engelke, U.F.H.; Rhee, K.; Williams, C.J.; Sundberg, J.P.; Wevers, R.A.; Tomlinson, R.E.; Jansen, R.S.; et al. The membrane protein ANKH is crucial for bone mechanical performance by mediating cellular export of citrate and ATP. PLoS Genet. 2020, 16, e1008884. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mycielska, M.E.; James, E.N.; Parkinson, E.K. Metabolic Alterations in Cellular Senescence: The Role of Citrate in Ageing and Age-Related Disease. Int. J. Mol. Sci. 2022, 23, 3652. https://doi.org/10.3390/ijms23073652
Mycielska ME, James EN, Parkinson EK. Metabolic Alterations in Cellular Senescence: The Role of Citrate in Ageing and Age-Related Disease. International Journal of Molecular Sciences. 2022; 23(7):3652. https://doi.org/10.3390/ijms23073652
Chicago/Turabian StyleMycielska, Maria Elzbieta, Emma Naomi James, and Eric Kenneth Parkinson. 2022. "Metabolic Alterations in Cellular Senescence: The Role of Citrate in Ageing and Age-Related Disease" International Journal of Molecular Sciences 23, no. 7: 3652. https://doi.org/10.3390/ijms23073652