
Genetic Inactivation of Free Fatty Acid Receptor 3 Impedes Behavioral Deficits and Pathological Hallmarks in the APPswe Alzheimer’s Disease Mouse Model

 , ,
, ,  and
and
Abstract
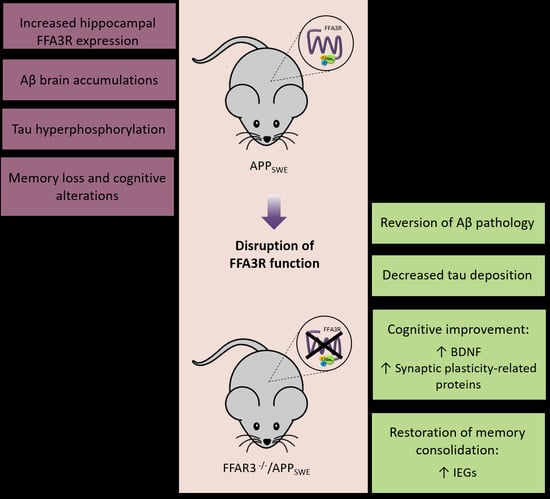
:
1. Introduction
2. Results
2.1. Expression of Transcripts for FFA Receptors in Postmortem Human Hippocampal Samples
2.2. Expression of Transcripts for FFA Receptors in the Hippocampus of WT and Transgenic Mice
2.3. Genetic Inactivation of FFA3R Reverses the Cognitive Impairment of APPswe Mice
2.4. Aβ Pathology Is Reversed in the Brain of FFA3R−/−/APPswe Mice
2.5. Hippocampus of FFA3R−/−/APPswe Mice Show a Decrease in Tau Pathology
2.6. FFA3R−/−/APPswe Mice Show an Increase in Mature BDNF and the Expression of Synaptic Plasticity-Related Proteins
3. Discussion
4. Materials and Methods
4.1. Human Brain Samples
4.2. Animals and Behavior Test
4.3. Morris Water Maze Test (MWM)
4.4. Fear Conditioning-Based Memory Induction Protocol
4.5. Brain Tissue Processing for Immunohistochemistry
4.6. Protein Extracts
4.7. Determination of Aβ Levels
4.8. Immunoblotting
4.9. RNA Extraction and Quantitative Reverse Transcription PCR (RT-qPCR)
4.10. Data Analysis and Statistical Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maccioni, R.B.; Muñoz, J.P.; Barbeito, L. The Molecular Bases of Alzheimer’s Disease and Other Neurodegenerative Disorders. Arch. Med. Res. 2001, 32, 367–381. [Google Scholar] [CrossRef]
- Di Domenico, F.; Sultana, R.; Barone, E.; Perluigi, M.; Cini, C.; Mancuso, C.; Cai, J.; Pierce, W.M.; Butterfield, D.A. Quantitative proteomics analysis of phosphorylated proteins in the hippocampus of Alzheimer’s disease subjects. J. Proteom. 2011, 74, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Bascoul-Colombo, C.; Guschina, I.A.; Maskrey, B.H.; Good, M.; O’Donnell, V.B.; Harwood, J.L. Dietary DHA supplementation causes selective changes in phospholipids from different brain regions in both wild type mice and the Tg2576 mouse model of Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Falomir-Lockhart, L.J.; Cavazzutti, G.F.; Giménez, E.; Toscani, A.M. Fatty acid signaling mechanisms in neural cells: Fatty acid receptors. Front. Cell. Neurosci. 2019, 13, 162. [Google Scholar]
- Fonteh, A.N.; Chiang, A.J.; Arakaki, X.; Edminster, S.P.; Harrington, M.G. Accumulation of Cerebrospinal Fluid Glycerophospholipids and Sphingolipids in Cognitively Healthy Participants With Alzheimer’s Biomarkers Precedes Lipolysis in the Dementia Stage. Front. Neurosci. 2020, 14, 1320. [Google Scholar] [CrossRef]
- Vitiello, G.; Di Marino, S.; D’Ursi, A.M.; D’Errico, G. Omega-3 Fatty Acids Regulate the Interaction of the Alzheimer’s Aβ(25–35) Peptide with Lipid Membranes. Langmuir 2013, 29, 14239–14245. [Google Scholar] [CrossRef] [Green Version]
- González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J.L. Combination of metabolomic and phospholipid-profiling approaches for the study of Alzheimer’s disease. J. Proteom. 2014, 104, 37–47. [Google Scholar] [CrossRef]
- Nagarathinam, A.; Höflinger, P.; Bühler, A.; Schafer, C.; McGovern, G.; Jeffrey, M.; Staufenbiel, M.; Jucker, M.; Baumann, F. Membrane-Anchored A Accelerates Amyloid Formation and Exacerbates Amyloid-Associated Toxicity in Mice. J. Neurosci. 2013, 33, 19284–19294. [Google Scholar] [CrossRef] [Green Version]
- Canale, C.; Seghezza, S.; Vilasi, S.; Carrotta, R.; Bulone, D.; Diaspro, A.; Biagio, P.L.S.; Dante, S. Different effects of Alzheimer’s peptide Aβ(1–40) oligomers and fibrils on supported lipid membranes. Biophys. Chem. 2013, 182, 23–29. [Google Scholar] [CrossRef]
- Shimohama, S.; Fujimoto, S.; Tresser, N.; Richey, P.; Perry, G.; Whitehouse, P.J.; Homma, Y.; Takenawa, T.; Taniguchi, T.; Suenaga, T.; et al. Aberrant Phosphoinositide Metabolism in Alzheimer’s Diseasea. Ann. N. Y. Acad. Sci. 1993, 695, 46–49. [Google Scholar] [CrossRef]
- Svennerholm, L.; Gottfries, C.-G. Membrane Lipids, Selectively Diminished in Alzheimer Brains, Suggest Synapse Loss as a Primary Event in Early-Onset Form (Type I) and Demyelination in Late-Onset Form (Type II). J. Neurochem. 2008, 62, 1039–1047. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Baron, R.; Babcock, A.A.; Nemirovsky, A.; Finsen, B.; Monsonego, A. Accelerated microglial pathology is associated with A β plaques in mouse models of A lzheimer’s disease. Aging Cell 2014, 13, 584–595. [Google Scholar] [CrossRef]
- Nichols, M.R.; St-Pierre, M.-K.; Wendeln, A.-C.; Makoni, N.J.; Gouwens, L.K.; Garrad, E.C.; Sohrabi, M.; Neher, J.J.; Tremblay, M.-E.; Combs, C.K. Inflammatory mechanisms in neurodegeneration. J. Neurochem. 2019, 149, 562–581. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.T.T.; Corsini, S.; Kellingray, L.; Hegarty, C.; Le Gall, G.; Narbad, A.; Müller, M.; Tejera, N.; O’Toole, P.W.; Minihane, A.-M.; et al. APOE genotype influences the gut microbiome structure and function in humans and mice: Relevance for Alzheimer’s disease pathophysiology. FASEB J. 2019, 33, 8221–8231. [Google Scholar] [CrossRef] [Green Version]
- Uauy, R.; Mena, P. Lipids and Neurodevelopment. Nutr. Rev. 2001, 59, S34–S48. [Google Scholar] [CrossRef]
- Martín, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marín, R.; Ferrer, I.; Díaz, M. Lipid Alterations in Lipid Rafts from Alzheimer’s Disease Human Brain Cortex. J. Alzheimer’s Dis. 2010, 19, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.W.; Braidy, N.; Poljak, A.; Pickford, R.; Thambisetty, M.; Sachdev, P.S. Dysregulation of lipids in Alzheimer’s disease and their role as potential biomarkers. Alzheimer’s Dement. 2017, 13, 810–827. [Google Scholar] [CrossRef]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef]
- Bazan, N.G. Lipid Signaling in Neural Plasticity, Brain Repair, and Neuroprotection. Mol. Neurobiol. 2005, 32, 089–104. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F. Role Of Lipids In Brain Injury And Diseases. Futur. Lipidol. 2007, 2, 403–422. [Google Scholar] [CrossRef] [Green Version]
- Yamashima, T.; Ota, T.; Mizukoshi, E.; Nakamura, H.; Yamamoto, Y.; Kikuchi, M.; Yamashita, T.; Kaneko, S. Intake of ω-6 Polyunsaturated Fatty Acid-Rich Vegetable Oils and Risk of Lifestyle Diseases. Adv. Nutr. Int. Rev. J. 2020, 11, 1489–1509. [Google Scholar] [CrossRef]
- Isacson, O.; Brekk, O.R.; Hallett, P.J. Novel Results and Concepts Emerging From Lipid Cell Biology Relevant to Degenerative Brain Aging and Disease. Front. Neurol. 2019, 10, 1053. [Google Scholar] [CrossRef] [PubMed]
- Marinissen, M.J.; Gutkind, J. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Zamarbide, M.; Etayo-Labiano, I.; Ricobaraza, A.; Martinez-Pinilla, E.; Aymerich, M.S.; Lanciego, J.; Pérez-Mediavilla, A.; Franco, R. GPR40 activation leads to CREB and ERK phosphorylation in primary cultures of neurons from the mouse CNS and in human neuroblastoma cells. Hippocampus 2014, 24, 733–739. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Chitre, N.M.; Moniri, N.H.; Murnane, K.S. Omega-3 Fatty Acids as Druggable Therapeutics for Neurodegenerative Disorders. CNS Neurol. Disord.-Drug Targets 2020, 18, 735–749. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors. J. Cereb. Blood Flow Metab. 2019, 176, S21–S141. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, L.A.; Smith, N.J.; Milligan, G. International Union of Pharmacology. LXXI. Free Fatty Acid Receptors FFA1, -2, and -3: Pharmacology and Pathophysiological Functions. Pharmacol. Rev. 2008, 60, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Tikhonova, I.G. Application of GPCR Structures for Modelling of Free Fatty Acid Receptors. In Free Fatty Acid Receptors; Springer: Cham, Switzerland, 2016; Volume 236, pp. 57–77. [Google Scholar] [CrossRef] [Green Version]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.-Y.; Lannoy, V.; Decobecq, M.-E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional Characterization of Human Receptors for Short Chain Fatty Acids and Their Role in Polymorphonuclear Cell Activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Curto, E.; Milligan, G. Metabolism meets immunity: The role of free fatty acid receptors in the immune system. Biochem. Pharmacol. 2016, 114, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.-T.; Leng, J.; Xie, Z.; Li, S.-L.; Zhao, W.; Tang, Q.-L. GPR40: A therapeutic target for mediating insulin secretion. Int. J. Mol. Med. 2012, 30, 1261–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.; Smith, N.J.; Milligan, G. Experimental Challenges to Targeting Poorly Characterized GPCRs: Uncovering the Therapeutic Potential for Free Fatty Acid Receptors. Adv. Pharmacol. 2011, 62, 175–218. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kimura, I.; Inoue, D.; Ichimura, A.; Hirasawa, A. Free Fatty Acid Receptors and Their Role in Regulation of Energy Metabolism. Rev. Physiol. Biochem. Pharmacol. 2013, 164, 77–116. [Google Scholar] [CrossRef]
- Poitout, V. Fatty Acids and Insulin Secretion: From FFAR and Near? Diabetes 2018, 67, 1932–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miletta, M.C.; Petkovic, V.; Eblé, A.; Ammann, R.A.; Flück, C.E.; Mullis, P.-E. Butyrate Increases Intracellular Calcium Levels and Enhances Growth Hormone Release from Rat Anterior Pituitary Cells via the G-Protein-Coupled Receptors GPR41 and 43. PLoS ONE 2014, 9, e107388. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Tian, X.; Xu, D.; Zheng, F.; Lu, X.; Zhang, Y.; Ma, Y.; Li, Y.; Xu, X.; Zhu, B.; et al. GPR40 modulates epileptic seizure and NMDA receptor function. Sci. Adv. 2018, 4, eaau2357. [Google Scholar] [CrossRef] [Green Version]
- Karki, P.; Kurihara, T.; Nakamachi, T.; Watanabe, J.; Asada, T.; Oyoshi, T.; Shioda, S.; Yoshimura, M.; Arita, K.; Miyata, A. Attenuation of inflammatory and neuropathic pain behaviors in mice through activation of free fatty acid receptor GPR40. Mol. Pain 2015, 11, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Nøhr, M.K.; Egerod, K.; Christiansen, S.; Gille, A.; Offermanns, S.; Schwartz, T.; Møller, M. Expression of the short chain fatty acid receptor GPR41/FFAR3 in autonomic and somatic sensory ganglia. Neuroscience 2015, 290, 126–137. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, E.A. Gut feelings: The emerging biology of gut–brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Jiménez-Pranteda, M.L.; Chilloux, J.; Brial, F.; Myridakis, A.; Aranias, T.; Magnan, C.; Gibson, G.R.; Sanderson, J.D.; Nicholson, J.K.; et al. Metabolic retroconversion of trimethylamine N-oxide and the gut microbiota. Microbiome 2018, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Van De Wouw, M.; Boehme, M.; Lyte, J.M.; Wiley, N.; Strain, C.; O’Sullivan, O.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 2018, 596, 4923–4944. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-F.; Shan, C.; Zhuang, S.-Y.; Zhuang, Q.-Q.; Ghosh, A.; Zhu, K.-C.; Kong, X.-K.; Wang, S.-M.; Gong, Y.-L.; Yang, Y.-Y.; et al. Gut microbiota-derived propionate mediates the neuroprotective effect of osteocalcin in a mouse model of Parkinson’s disease. Microbiome 2021, 9, 34. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, W.; Chen, L.; Qiu, X.; Li, C.; Huang, C.-X.; Gong, X.; Min, Q.-C.; Lu, F.; Wan, J.-Y.; et al. The early changes in behavior and the myelinated fibers of the white matter in the Tg2576 transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2013, 555, 112–117. [Google Scholar] [CrossRef]
- Avila, J.; Perry, G. A Multilevel View of the Development of Alzheimer’s Disease. Neuroscience 2020, 457, 283–293. [Google Scholar] [CrossRef]
- Rabbito, A.; Dulewicz, M.; Kulczyńska-Przybik, A.; Mroczko, B. Biochemical Markers in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1989. [Google Scholar] [CrossRef] [Green Version]
- Stoiljkovic, M.; Horvath, T.L.; Hajós, M. Therapy for Alzheimer’s disease: Missing targets and functional markers? Ageing Res. Rev. 2021, 68, 101318. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G Protein-coupled Receptors GPR41 and GPR43 Are Activated by Propionate and Other Short Chain Carboxylic Acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couto, M.R.; Gonçalves, P.; Magro, F.; Martel, F. Microbiota-derived butyrate regulates intestinal inflammation: Focus on inflammatory bowel disease. Pharmacol. Res. 2020, 159, 104947. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Bauer, B.; Abner, E.L.; Ashkenazy-Frolinger, T.; Hartz, A.M.S. A Comprehensive Behavioral Test Battery to Assess Learning and Memory in 129S6/Tg2576 Mice. PLoS ONE 2016, 11, e0147733. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Chang, K.-A.; Ha, T.-Y.; Kim, J.; Ha, S.; Shin, K.-Y.; Moon, C.; Nacken, W.; Kim, H.-S.; Suh, Y.-H. S100A9 Knockout Decreases the Memory Impairment and Neuropathology in Crossbreed Mice of Tg2576 and S100A9 Knockout Mice Model. PLoS ONE 2014, 9, e88924. [Google Scholar] [CrossRef]
- Lanahan, A.; Worley, P. Immediate-Early Genes and Synaptic Function. Neurobiol. Learn. Mem. 1998, 70, 37–43. [Google Scholar] [CrossRef]
- Guzowski, J.F. Insights into immediate-early gene function in hippocampal memory consolidation using antisense oligonucleotide and fluorescent imaging approaches. Hippocampus 2002, 12, 86–104. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Thomsen, M.S.; Mikkelsen, J.D. Reduced basal and novelty-induced levels of activity-regulated cytoskeleton associated protein (Arc) and c-Fos mRNA in the cerebral cortex and hippocampus of APPswe/PS1ΔE9 transgenic mice. Neurochem. Int. 2013, 63, 54–60. [Google Scholar] [CrossRef]
- Sierksma, A.; Hove, D.V.D.; Pfau, F.; Philippens, M.; Bruno, O.; Fedele, E.; Ricciarelli, R.; Steinbusch, H.; Vanmierlo, T.; Prickaerts, J. Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology 2014, 77, 120–130. [Google Scholar] [CrossRef]
- Palop, J.; Chin, J.; Bien-Ly, N.; Massaro, C.; Yeung, B.Z.; Yu, G.-Q.; Mucke, L. Vulnerability of Dentate Granule Cells to Disruption of Arc Expression in Human Amyloid Precursor Protein Transgenic Mice. J. Neurosci. 2005, 25, 9686–9693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reese, L.C.; Laezza, F.; Woltjer, R.; Taglialatela, G. Dysregulated phosphorylation of Ca2+/calmodulin-dependent protein kinase II-α in the hippocampus of subjects with mild cognitive impairment and Alzheimer’s disease. J. Neurochem. 2011, 119, 791–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Gylys, K.H.; Fein, J.A.; Yang, F.; Wiley, D.J.; Miller, C.A.; Cole, G.M. Synaptic Changes in Alzheimer’s Disease: Increased Amyloid-β and Gliosis in Surviving Terminals Is Accompanied by Decreased PSD-95 Fluorescence. Am. J. Pathol. 2004, 165, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ Influx Regulates BDNF Transcription by a CREB Family Transcription Factor-Dependent Mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [Green Version]
- Seidah, N.G.; Chrétien, M. Proprotein and prohormone convertases: A family of subtilases generating diverse bioactive polypeptides1Published on the World Wide Web on 17 August 1999.1. Brain Res. 1999, 848, 45–62. [Google Scholar] [CrossRef]
- Bibel, M.; Barde, Y.-A. Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000, 14, 2919–2937. [Google Scholar] [CrossRef] [Green Version]
- Schindowski, K.; Belarbi, K.; Buée, L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav. 2008, 7 (Suppl. S1), 43–56. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Hobson, V.; Hall, J.R.; Waring, S.C.; Chan, W.; Massman, P.; Lacritz, L.; Cullum, C.M.; Diaz-Arrastia, R. Brain-Derived Neurotrophic Factor Levels in Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 17, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Holsinger, R.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in Alzheimer’s disease. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.-F.; Kuo, Y.-M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble Amyloid β Peptide Concentration as a Predictor of Synaptic Change in Alzheimer’s Disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Vbeyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Aβ amyloid as a determinant of severity of neuro-degeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Belbin, O.; Zou, F.; Allen, M.; Ertekin-Taner, N.; Ansari, M.; Wilcox, S.L.; Kashino, M.R.; Ma, L.; Younkin, L.H.; et al. Concordant Association of Insulin Degrading Enzyme Gene (IDE) Variants with IDE mRNA, Aß, and Alzheimer’s Disease. PLoS ONE 2010, 5, e8764. [Google Scholar] [CrossRef]
- Kilger, E.; Buehler, A.; Woelfing, H.; Kumar, S.; Kaeser, S.A.; Nagarathinam, A.; Walter, J.; Jucker, M.; Coomaraswamy, J. BRI2 Protein Regulates β-Amyloid Degradation by Increasing Levels of Secreted Insulin-degrading Enzyme (IDE)*. J. Biol. Chem. 2011, 286, 37446–37457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.-P.; Qian, X.; Xie, Y.; Qi, Y.; Peng, M.-F.; Zhan, B.-C.; Lou, Z.-Q. Betaine suppressed Aβ generation by altering amyloid precursor protein processing. Neurol. Sci. 2014, 35, 1009–1013. [Google Scholar] [CrossRef]
- Probst; Botez; Tolnay Neuropathology of Alzheimer’s disease. Ther. Umsch. 1999, 56, 88–93. [CrossRef]
- Giannakopoulos, P.; Kövari, E.; Gold, G.; von Gunten, A.; Hof, P.R.; Bouras, C. Pathological Substrates of Cognitive Decline in Alzheimer’s Disease. Front. Neurol. Neurosci. 2009, 24, 20–29. [Google Scholar] [CrossRef]
- Pooler, A.M.; Noble, W.; Hanger, D.P. A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology 2014, 76, 1–8. [Google Scholar] [CrossRef]
- Ferrer, I. Stress kinases involved inTau phosphorylation in alzheimer’s disease, tauopathies and APP transgenic mice. Neurotox. Res. 2004, 6, 469–475. [Google Scholar] [CrossRef]
- Chauhan, N.B.; Siegel, G.J.; Feinstein, D.L. Propentofylline attenuates tau hyperphosphorylation in Alzheimer’s Swedish mutant model Tg2576. Neuropharmacology 2005, 48, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Gomez-Isla, T.; Puig, B.; Freixes, M.; Ribe, E.; Dalfo, E.; Avila, J. Current Advances on Different Kinases Involved in Tau Phosphorylation, and Implications in Alzheimers Disease and Tauopathies. Curr. Alzheimer Res. 2005, 2, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamarbide, M.; Gil-Bea, F.J.; Bannenberg, P.; Martinez-Pinilla, E.; Sandoval, J.; Franco, R.; Pérez-Mediavilla, A. Maternal imprinting on cognition markers of wild type and transgenic Alzheimer’s disease model mice. Sci. Rep. 2018, 8, 6434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado-Tejedor, M.; Hervias, I.; Ricobaraza, A.; Puerta, E.; Pérez-Roldán, J.; García-Barroso, C.; Franco, R.; Aguirre, N.; García-Osta, A. Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2011, 164, 2029–2041. [Google Scholar] [CrossRef] [Green Version]
- Dunah, A.W.; Wang, Y.; Yasuda, R.P.; Kameyama, K.; Huganir, R.L.; Wolfe, B.B.; Standaert, D.G. Alterations in subunit expression, composition, and phosphorylation of striataln-methyl-d-aspartate glutamate receptors in a rat 6-hydroxydopamine model of Parkinson’s disease. Mol. Pharmacol. 2000, 57, 342–352. [Google Scholar]
- Milligan, G.; Shimpukade, B.; Ulven, T.; Hudson, B.D. Complex Pharmacology of Free Fatty Acid Receptors. Chem. Rev. 2016, 117, 67–110. [Google Scholar] [CrossRef]
- Milligan, G.; Bolognini, D.; Sergeev, E. Ligands at the Free Fatty Acid Receptors 2/3 (GPR43/GPR41). Free. Fat. Acid Recept. 2016, 236, 17–32. [Google Scholar] [CrossRef]
- Milligan, G.; Stoddart, L.; Smith, N.J. Agonism and allosterism: The pharmacology of the free fatty acid receptors FFA2 and FFA3. J. Cereb. Blood Flow Metab. 2009, 158, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Grundmann, M.; Bender, E.; Schamberger, J.; Eitner, F. Pharmacology of Free Fatty Acid Receptors and Their Allosteric Modulators. Int. J. Mol. Sci. 2021, 22, 1763. [Google Scholar] [CrossRef]
- Watterson, K.R.; Hudson, B.; Ulven, T.; Emilligan, G. Treatment of Type 2 Diabetes by Free Fatty Acid Receptor Agonists. Front. Endocrinol. 2014, 5, 137. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Braak Stage | Cases (n) | M/F | Age *, Year | PMD *, h |
|---|---|---|---|---|
| ND | 6 | 4/2 | 35.5 (19–54) | 3.2 (2.15–6) |
| Braak III–IV | 4 | 2/2 | 83.5 (76–89) | 2.5 (2–3.37) |
| Braak V | 4 | 0/4 | 87 (77–92) | 2.3 |
| Braak VI | 6 | 1/5 | 84.2 (79–89) | 2.3 (1.25–3.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamarbide, M.; Martinez-Pinilla, E.; Gil-Bea, F.; Yanagisawa, M.; Franco, R.; Perez-Mediavilla, A. Genetic Inactivation of Free Fatty Acid Receptor 3 Impedes Behavioral Deficits and Pathological Hallmarks in the APPswe Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2022, 23, 3533. https://doi.org/10.3390/ijms23073533
Zamarbide M, Martinez-Pinilla E, Gil-Bea F, Yanagisawa M, Franco R, Perez-Mediavilla A. Genetic Inactivation of Free Fatty Acid Receptor 3 Impedes Behavioral Deficits and Pathological Hallmarks in the APPswe Alzheimer’s Disease Mouse Model. International Journal of Molecular Sciences. 2022; 23(7):3533. https://doi.org/10.3390/ijms23073533
Chicago/Turabian StyleZamarbide, Marta, Eva Martinez-Pinilla, Francisco Gil-Bea, Masashi Yanagisawa, Rafael Franco, and Alberto Perez-Mediavilla. 2022. "Genetic Inactivation of Free Fatty Acid Receptor 3 Impedes Behavioral Deficits and Pathological Hallmarks in the APPswe Alzheimer’s Disease Mouse Model" International Journal of Molecular Sciences 23, no. 7: 3533. https://doi.org/10.3390/ijms23073533