
The Role of Endoplasmic Reticulum Stress and NLRP3 Inflammasome in Liver Disorders

 , and
, and
Abstract
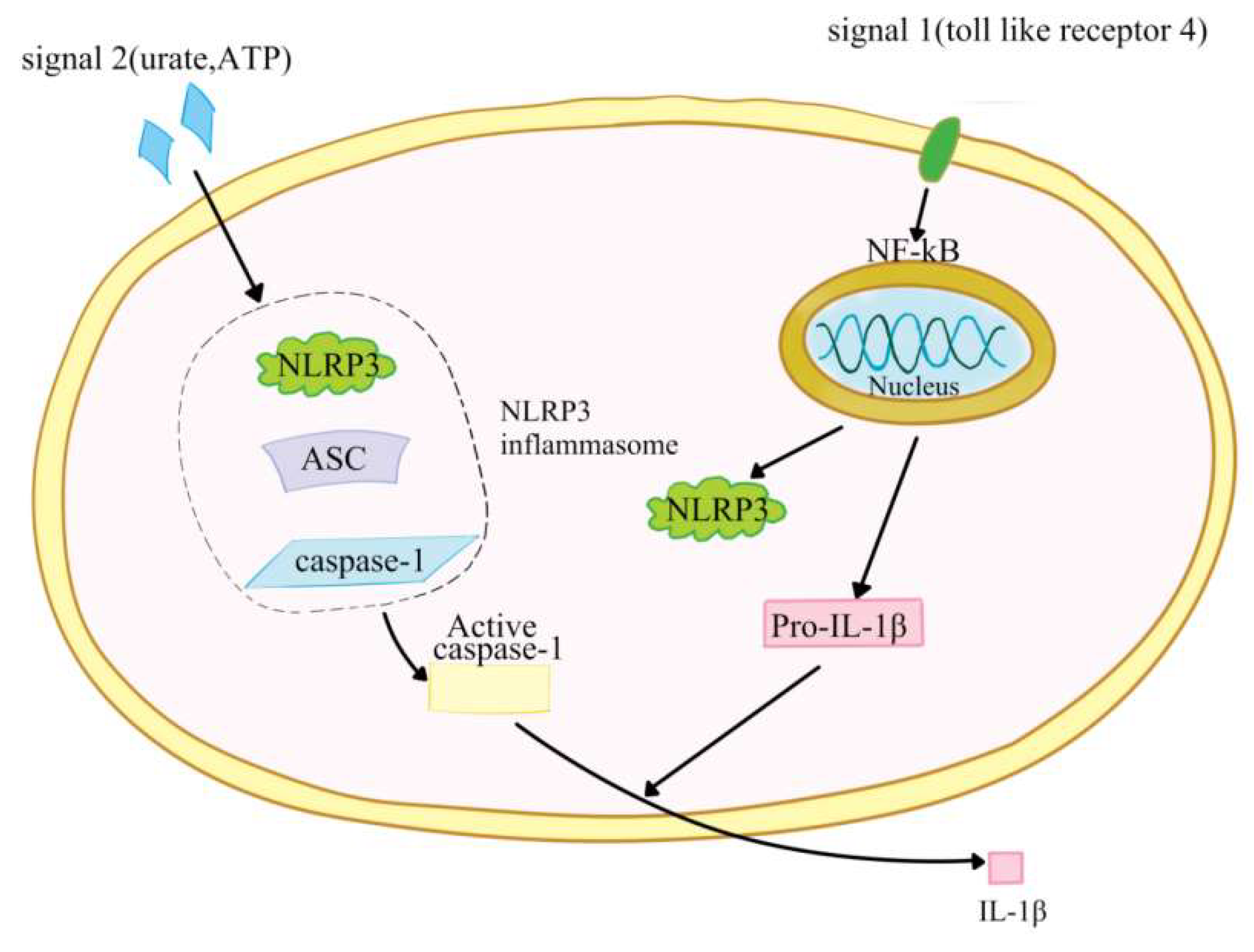
:1. Introduction
2. The Role of Endoplasmic Reticulum Stress and the NLRP3 Inflammasome in Nonalcoholic Fatty Liver Disease
2.1. The Role of Endoplasmic Reticulum Stress and NLRP3 Inflammasome in Nonalcoholic Steatohepatitis
2.2. Bax Inhibitor-1 Improves NAFLD through Endoplasmic Reticulum Stress and NLRP3 Inflammasome
2.3. Ginsenoside Rg1 Improves NAFLD through Endoplasmic Reticulum Stress and NLRP3 Inflammasome
2.4. Acetylantroquinonol B Improves NAFLD through Endoplasmic Reticulum Stress and the NLRP3 Inflammasome
3. The Role of Endoplasmic Reticulum Stress and the NLRP3 Inflammasome in Hepatic Ischemia–Reperfusion
4. The Role of Endoplasmic Reticulum Stress and the NLRP3 Inflammasome in Hepatotoxicity
4.1. Allicin Improves Hepatotoxicity through Endoplasmic Reticulum Stress and the NLRP3 Inflammasome
4.2. Baicalin Improves Hepatotoxicity through Endoplasmic Reticulum Stress and NLRP3 Inflammasome 13
5. Farnesoid X Receptor Improves Liver Injury through Endoplasmic Reticulum Stress and NLRP3 Inflammasome
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Volt, H.; Garcia, J.A.; Doerrier, C.; Diaz-Casado, M.E.; Guerra-Librero, A.; Lopez, L.C.; Escames, G.; Tresguerres, J.A.; Acuna-Castroviejo, D. Same molecule but different expression: Aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J. Pineal Res. 2016, 60, 193–205. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Nunez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Wang, N.; Qiu, T.; Sun, X. The Role of Autophagy and NLRP3 Inflammasome in Liver Fibrosis. Biomed Res. Int. 2020, 2020, 7269150. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Kinio, A.; Saleh, M. Functions of NOD-Like Receptors in Human Diseases. Front. Immunol. 2013, 4, 333. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.X.; Halfmann, R.; Chen, Z.J. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Li, X.; Wang, H. The Role of the Effects of Endoplasmic Reticulum Stress on NLRP3 Inflammasome in Diabetes. Front. Cell Dev. Biol. 2021, 9, 663528. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Junkins, R.D.; Kurkjian, C.J.; Holley-Guthrie, E.; Pendse, A.A.; El Morabiti, R.; Petrucelli, A.; Barber, G.N.; Benedict, C.A.; Ting, J.P. A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med. 2017, 214, 3611–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolova, M.; Ranheim, T.; Louwe, M.C.; Halvorsen, B.; Yndestad, A.; Aukrust, P. NLRP3 Inflammasome: A Novel Player in Metabolically Induced Inflammation-Potential Influence on the Myocardium. J. Cardiovasc. Pharmacol. 2019, 74, 276–284. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; McGeough, M.D.; Inzaugarat, M.E.; Eguchi, A.; Schuster, S.; Johnson, C.D.; Pena, C.A.; Geisler, L.J.; Papouchado, B.G.; Hoffman, H.M.; et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 2018, 67, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Akter, A.; Hossain, S.; Sarker, T.; Safa, S.A.; Mustafa, Q.G.; Muhammad, S.A.; Munir, F. Role of NLRP3 inflammasome in liver disease. J. Dig. Dis. 2020, 21, 430–436. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Karnovsky, A.; Sans, M.D.; Andrews, P.C.; Williams, J.A. Molecular characterization of the endoplasmic reticulum: Insights from proteomic studies. Proteomics 2010, 10, 4040–4052. [Google Scholar] [CrossRef]
- Mandl, J.; Meszaros, T.; Banhegyi, G.; Csala, M. Minireview: Endoplasmic reticulum stress: Control in protein, lipid, and signal homeostasis. Mol. Endocrinol. 2013, 27, 384–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel-Aldao, R. The unfolded protein response, inflammation, oscillators, and disease: A systems biology approach. Endoplasmic Reticulum Stress Dis. 2015, 2, 30–52. [Google Scholar] [CrossRef]
- McCaffrey, K.; Braakman, I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016, 60, 227–235. [Google Scholar] [CrossRef]
- So, J.S. Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol. Cells 2018, 41, 705–716. [Google Scholar] [CrossRef]
- Ji, T.; Han, Y.; Yang, W.; Xu, B.; Sun, M.; Jiang, S.; Yu, Y.; Jin, Z.; Ma, Z.; Yang, Y.; et al. Endoplasmic reticulum stress and NLRP3 inflammasome: Crosstalk in cardiovascular and metabolic disorders. J. Cell. Physiol. 2019, 234, 14773–1478821. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef]
- Oyadomari, S.; Harding, H.P.; Zhang, Y.; Oyadomari, M.; Ron, D. Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008, 7, 520–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [Green Version]
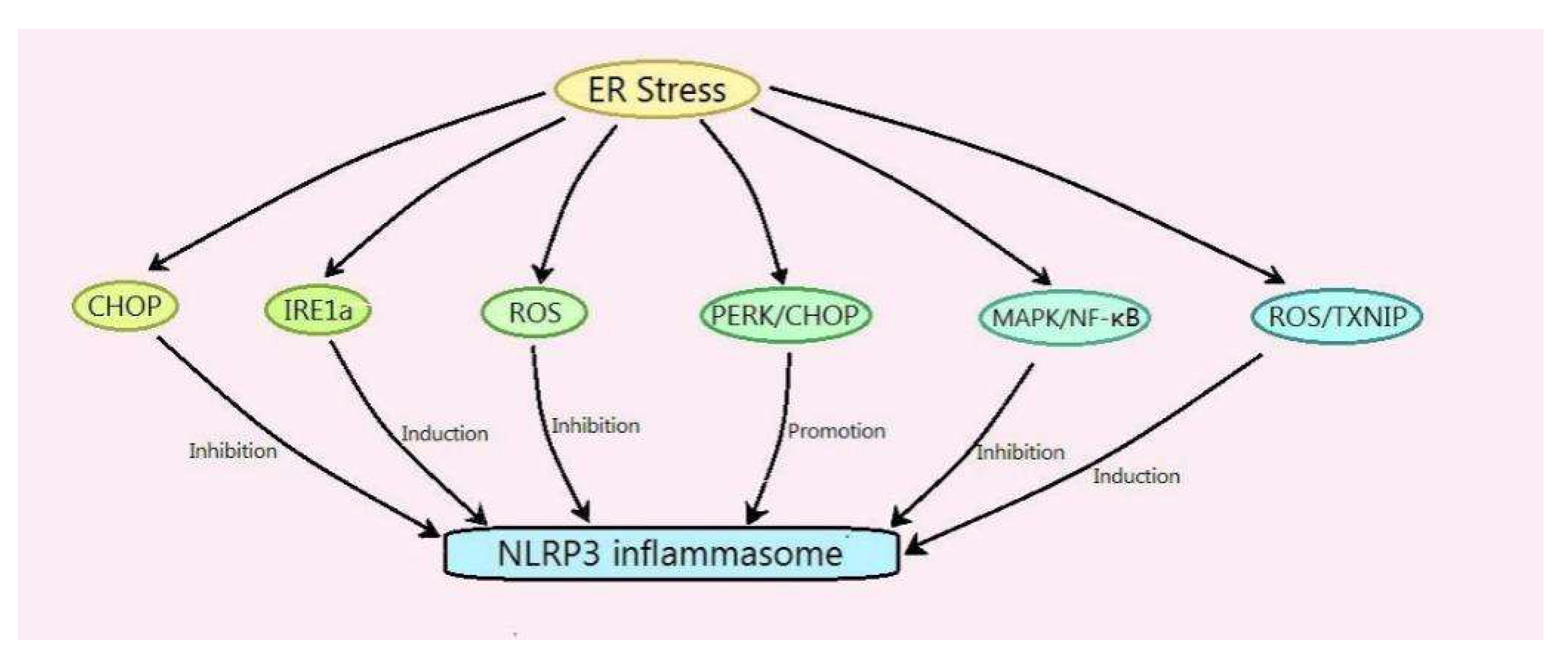
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Jin, C.; Wu, Y.; Zhang, Y.; Wang, X.; Huang, W.; Li, J.; Wu, S.; Gao, X. Prospective study of perceived dietary salt intake and the risk of non-alcoholic fatty liver disease. J. Hum. Nutr. Diet. 2019, 32, 802–809. [Google Scholar] [CrossRef] [PubMed]
- DeWeerdt, S. Disease progression: Divergent paths. Nature 2017, 551, S92–S93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Xu, C.; Shi, J.; Ding, J.; Wan, X.; Chen, D.; Gao, J.; Li, C.; Zhang, J.; Lin, Y.; et al. Fatty acids promote fatty liver disease via the dysregulation of 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Gut 2018, 67, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Issa, D.; Patel, V.; Sanyal, A.J. Future therapy for non-alcoholic fatty liver disease. Liver Int. 2018, 38 (Suppl. S1), 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; Diehl, A.M. Nonalcoholic Steatohepatitis. Annu. Rev. Med. 2017, 68, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.W.; Wang, Z.M.; Sun, S.M.; Su, Y.; Li, Z.H.; Shao, J.J.; Tan, S.Z.; Chen, A.P.; Wang, S.J.; Zhang, Z.L.; et al. Endoplasmic reticulum stress and protein degradation in chronic liver disease. Pharmacol. Res. 2020, 161, 105218. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef] [Green Version]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; He, Y.; Ming, H.; Lei, S.; Leng, Y.; Xia, Z.Y. Lipopolysaccharide (LPS) Aggravates High Glucose- and Hypoxia/Reoxygenation-Induced Injury through Activating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis in H9C2 Cardiomyocytes. J. Diabetes Res. 2019, 2019, 8151836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly-Maitre, B.; Belgardt, B.F.; Jordan, S.D.; Coornaert, B.; von Freyend, M.J.; Kleinridders, A.; Mauer, J.; Cuddy, M.; Kress, C.L.; Willmes, D.; et al. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J. Biol. Chem. 2010, 285, 6198–6207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisbona, F.; Rojas-Rivera, D.; Thielen, P.; Zamorano, S.; Todd, D.; Martinon, F.; Glavic, A.; Kress, C.; Lin, J.H.; Walter, P.; et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1α. Mol. Cell 2009, 33, 679–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaupin, C.; Vallee, D.; Rousseau, D.; Patouraux, S.; Bonnafous, S.; Adam, G.; Luciano, F.; Luci, C.; Anty, R.; Iannelli, A.; et al. Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology 2018, 68, 515–532. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Gu, D.; Peng, J.; Jiang, K.; Li, Z.; Shi, J.; Yang, S.; Li, S.; Fan, X. Ginsenoside Rg1 Regulates Liver Lipid Factor Metabolism in NAFLD Model Rats. ACS Omega 2020, 5, 10878–10890. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Jeon, J.Y.; Yang, W.S.; Kim, C.H.; Eom, D.W. Combined Amelioration of Ginsenoside (Rg1, Rb1, and Rg3)-enriched Korean Red Ginseng and Probiotic Lactobacillus on Non-alcoholic Fatty Liver Disease. Curr. Pharm. Biotechnol. 2019, 20, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Yi, H.; Jiang, K.; Fakhar, S.H.; Shi, J.; He, Y.; Liu, B.; Guo, Y.; Fan, X.; Li, S. Transcriptome analysis reveals the efficacy of ginsenoside-Rg1 in the treatment of nonalcoholic fatty liver disease. Life Sci. 2021, 267, 118986. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, C.; Zhang, S.; Li, J.; Xiao, Q.; Huang, W. Ginsenoside Rg1 Protects against Non-alcoholic Fatty Liver Disease by Ameliorating Lipid Peroxidation, Endoplasmic Reticulum Stress, and Inflammasome Activation. Biol. Pharm. Bull. 2018, 41, 1638–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Ou, C.H.; Yen, I.C.; Lee, S.Y. 4-Acetylantroquinonol B Inhibits Osteoclastogenesis by Inhibiting the Autophagy Pathway in a Simulated Microgravity Model. Int. J. Mol. Sci. 2020, 21, 6971. [Google Scholar] [CrossRef]
- Yen, I.C.; Tu, Q.W.; Chang, T.C.; Lin, P.H.; Li, Y.F.; Lee, S.Y. 4-Acetylantroquinonol B ameliorates nonalcoholic steatohepatitis by suppression of ER stress and NLRP3 inflammasome activation. Biomed. Pharmacother. 2021, 138, 111504. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Schenk, S.; Imamura, T.; Babendure, J.L.; Sonoda, N.; Bae, E.J.; Oh, D.Y.; Lu, M.; Milne, J.C.; Westphal, C.; et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E419–E428. [Google Scholar] [CrossRef] [Green Version]
- Ding, R.B.; Bao, J.; Deng, C.X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Giles, A.; Nakamura, K.; Lee, J.W.; Hou, X.; Donmez, G.; Li, J.; Luo, Z.; Walsh, K.; et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 2011, 25, 1664–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.Q.; Chen, L.L.; Li, N.X. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 2007, 27, 708–715. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Li, X.; Xing, D.; Du, X.; Wang, Z.; Liu, G.; Li, X. Nobiletin alleviates palmitic acidinduced NLRP3 inflammasome activation in a sirtuin 1dependent manner in AML12 cells. Mol. Med. Rep. 2018, 18, 5815–5822. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhang, M.; Lu, J.; Zhang, X.; Xiong, Q.; Xu, Y.; Bao, Y.; Jia, W. Osteocalcin improves nonalcoholic fatty liver disease in mice through activation of Nrf2 and inhibition of JNK. Endocrine 2016, 53, 701–709. [Google Scholar] [CrossRef]
- Chambel, S.S.; Santos-Goncalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. Biomed. Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.W.; Zhao, G.J.; Li, X.L.; Hong, G.L.; Li, M.F.; Qiu, Q.M.; Wu, B.; Lu, Z.Q. SIRT1 exerts protective effects against paraquat-induced injury in mouse type II alveolar epithelial cells by deacetylating NRF2 in vitro. Int. J. Mol. Med. 2016, 37, 1049–1058. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Yao, W.; Chen, C.; Chen, H.; Huang, F.; Liu, Y.; Cai, J.; Yuan, D.; Hei, Z. Connexin 32 deficiency protects the liver against ischemia/reperfusion injury. Eur. J. Pharmacol. 2020, 876, 173056. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, T.N.; Victorino, J.P.; Bistafa Liu, J.; Tofoli Queiroz Campos, D.; Graf, C.; Jordani, M.C.; Carneiro d’Albuquerque, L.A.; Mendes, K.D.S.; Castro, E.S.O. Effect of Hepatic Preconditioning with the Use of Methylene Blue on the Liver of Wistar Rats Submitted to Ischemia and Reperfusion. Transplant. Proc. 2018, 50, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Petrowsky, H.; Hong, J.C.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Zhang, R.; Gu, J.; Ju, Z.; Wu, X.; Wang, Q.; Peng, H.; Qiu, J.; Zhou, J.; Cheng, F.; et al. Acidic Microenvironment Regulates the Severity of Hepatic Ischemia/Reperfusion Injury by Modulating the Generation and Function of Tregs via the PI3K-mTOR Pathway. Front. Immunol. 2019, 10, 2945. [Google Scholar] [CrossRef]
- Ramazani, E.; Akaberi, M.; Emami, S.A.; Tayarani-Najaran, Z. Biological and Pharmacological Effects of Gamma-oryzanol: An Updated Review of the Molecular Mechanisms. Curr. Pharm. Des. 2021, 27, 2299–2316. [Google Scholar] [CrossRef] [PubMed]
- Minatel, I.O.; Francisqueti, F.V.; Correa, C.R.; Lima, G.P. Antioxidant Activity of gamma-Oryzanol: A Complex Network of Interactions. Int. J. Mol. Sci. 2016, 17, 1107. [Google Scholar] [CrossRef] [Green Version]
- Szczesniak, K.A.; Ostaszewski, P.; Ciecierska, A.; Sadkowski, T. Investigation of nutriactive phytochemical-gamma-oryzanol in experimental animal models. J. Anim. Physiol. Anim. Nutr. 2016, 100, 601–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Zhong, F.; Cheng, H.; Li, T.; Chen, Y.; Tan, P.; Huang, M.; Liang, T.; Liu, Y.; Xia, X.; et al. The Dietary Supplement gamma-Oryzanol Attenuates Hepatic Ischemia Reperfusion Injury via Inhibiting Endoplasmic Reticulum Stress and HMGB1/NLRP3 Inflammasome. Oxid. Med. Cell. Longev. 2021, 2021, 4628050. [Google Scholar] [CrossRef]
- Katwal, G.; Baral, D.; Fan, X.; Weiyang, H.; Zhang, X.; Ling, L.; Xiong, Y.; Ye, Q.; Wang, Y. SIRT3 a Major Player in Attenuation of Hepatic Ischemia-Reperfusion Injury by Reducing ROS via Its Downstream Mediators: SOD2, CYP-D, and HIF-1alpha. Oxid. Med. Cell. Longev. 2018, 2018, 2976957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, R.C.; Lu, S.Z.; Luo, Y.; Wang, T.; Liang, H.; Zeng, J.; Liu, J.; Hu, H.X. Calpain silencing alleviates myocardial ischemia-reperfusion injury through the NLRP3/ASC/Caspase-1 axis in mice. Life Sci. 2019, 233, 116631. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, X.; Chen, P.; Li, Y.; Liu, S.; Liu, Q.; Zhang, H.; Wu, Z.; Song, K.; Liu, J.; et al. The ER stress sensor inositol-requiring enzyme 1alpha in Kupffer cells promotes hepatic ischemia-reperfusion injury. J. Biol. Chem. 2022, 298, 101532. [Google Scholar] [CrossRef]
- Sansano, M.; Heredia, A.; Peinado, I.; Andres, A. Dietary acrylamide: What happens during digestion. Food Chem. 2017, 237, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Rifai, L.; Saleh, F.A. A Review on Acrylamide in Food: Occurrence, Toxicity, and Mitigation Strategies. Int. J. Toxicol. 2020, 39, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Zucca, P.; Orhan, I.E.; Azzini, E.; Adetunji, C.O.; Mohammed, S.A.; Banerjee, S.K.; Sharopov, F.; Rigano, D.; Sharifi-Rad, J.; et al. Allicin and health: A comprehensive review. Trends Food Sci. Technol. 2019, 86, 502–516. [Google Scholar] [CrossRef]
- Shi, X.; Zhou, X.; Chu, X.; Wang, J.; Xie, B.; Ge, J.; Guo, Y.; Li, X.; Yang, G. Allicin Improves Metabolism in High-Fat Diet-Induced Obese Mice by Modulating the Gut Microbiota. Nutrients 2019, 11, 2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; He, X.; Sheng, Y.; Yang, C.; Xu, J.; Zheng, S.; Liu, J.; Xu, W.; Luo, Y.; Huang, K. Allicin-induced host-gut microbe interactions improves energy homeostasis. FASEB J. 2020, 34, 10682–10698. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Nan, B.; Wu, X.; Yan, H.; Yuan, Y. Allicin alleviates acrylamide-induced oxidative stress in BRL-3A cells. Life Sci. 2019, 231, 116550. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Chen, D.Y.; Liu, H.Y.; Yan, H.Y.; Yuan, Y. Protective effect of allicin against glycidamide-induced toxicity in male and female mice. Gen. Physiol. Biophys. 2015, 34, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Nan, B.; Yang, C.; Li, L.; Ye, H.; Yan, H.; Wang, M.; Yuan, Y. Allicin alleviated acrylamide-induced NLRP3 inflammasome activation via oxidative stress and endoplasmic reticulum stress in Kupffer cells and SD rats liver. Food Chem. Toxicol. 2021, 148, 111937. [Google Scholar] [CrossRef]
- Panahi, G.; Pasalar, P.; Zare, M.; Rizzuto, R.; Meshkani, R. High glucose induces inflammatory responses in HepG2 cells via the oxidative stress-mediated activation of NF-kappaB, and MAPK pathways in HepG2 cells. Arch. Physiol. Biochem. 2018, 124, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Ma, Y.; Tong, X.; Yang, W.; Dai, Y.; Pan, Y.; Ren, P.; Liu, L.; Fan, H.Y.; Zhang, Y.; et al. Metformin inhibits testosterone-induced endoplasmic reticulum stress in ovarian granulosa cells via inactivation of p38 MAPK. Hum. Reprod. 2020, 35, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Sun, C.; Han, C.; Han, N.; Zhang, M.; Li, G. Endoplasmic reticulum stress pathway PERK-eIF2α confers radioresistance in oropharyngeal carcinoma by activating NF-kB. Cancer Sci. 2017, 108, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Liu, Y.; Zhang, C. Pharmacokinetics and Bioavailability Enhancement of Baicalin: A Review. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 159–168. [Google Scholar] [CrossRef]
- Yu, H.; Chen, B.; Ren, Q. Baicalin relieves hypoxia-aroused H9c2 cell apoptosis by activating Nrf2/HO-1-mediated HIF1alpha/BNIP3 pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3657–3663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.T.; Wang, S.Q.; Su, J.; Xu, L.X.; Ji, Z.Y.; Zhang, R.Y.; Zhao, Q.W.; Ma, Z.Q.; Deng, X.Y.; Ma, S.P. Baicalin ameliorates neuroinflammation-induced depressive-like behavior through inhibition of toll-like receptor 4 expression via the PI3K/AKT/FoxO1 pathway. J. Neuroinflamm. 2019, 16, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Wang, Y.; Fan, Y.; Li, H.; Wang, C.; Zhang, J.; Zhang, S.; Han, X.; Wen, C. Lipidomics revealed idiopathic pulmonary fibrosis-induced hepatic lipid disorders corrected with treatment of baicalin in a murine model. AAPS J. 2015, 17, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Liu, M.; Yu, H.; Li, J.; Wang, S.; Zhang, Y.; Qiu, F.; Wang, T. Scutellaria baicalensis regulates FFA metabolism to ameliorate NAFLD through the AMPK-mediated SREBP signaling pathway. J. Nat. Med. 2018, 72, 655–666. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Deng, X.; Zhang, Y.; Xu, K. Baicalin protects AML-12 cells from lipotoxicity via the suppression of ER stress and TXNIP/NLRP3 inflammasome activation. Chem. Biol. Interact. 2017, 278, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Li, T.; Tang, Z.H.; Chang, L.L.; Zhu, H.; Chen, X.P.; Wang, Y.T.; Lu, J.J. Baicalein Triggers Autophagy and Inhibits the Protein Kinase B/Mammalian Target of Rapamycin Pathway in Hepatocellular Carcinoma HepG2 Cells. Phytother. Res. 2015, 29, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Mai, W.; Xu, Y.; Xu, J.; Zhao, D.; Ye, L.; Yu, G.; Wang, Z.; Lu, Q.; Lin, J.; Yang, T.; et al. Berberine Inhibits Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome Activation and Pyroptosis in Nonalcoholic Steatohepatitis via the ROS/TXNIP Axis. Front. Pharmacol. 2020, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Zheng, T.; Yang, X.; Li, W.; Wang, Q.; Chen, L.; Wu, D.; Bian, F.; Xing, S.; Jin, S. Salidroside Attenuates High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease via AMPK-Dependent TXNIP/NLRP3 Pathway. Oxid. Med. Cell. Longev. 2018, 2018, 8597897. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.H.; Chen, X.Y.; Hu, Q.H.; Wang, M.X.; Jin, R.; Zhang, Q.Y.; Wang, W.; Wang, R.; Kang, L.L.; et al. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid. Redox Signal. 2015, 22, 848–870. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, Z.; Feng, Q.; Chen, W.D.; Wang, Y.D. Farnesoid X receptor: A potential therapeutic target in multiple organs. Histol. Histopathol. 2020, 35, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.; Amar, M.J.; Guo, G.; Brewer, H.B., Jr.; Gonzalez, F.J.; Sinal, C.J. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J. Biol. Chem. 2003, 278, 2563–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adorini, L.; Pruzanski, M.; Shapiro, D. Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov. Today 2012, 17, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62, S25–S37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, R.M.; Reid, A.E. FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr. Atheroscler. Rep. 2015, 17, 500. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Rho, H.S.; Kim, A.; Kim, T.H.; Jang, K.; Jun, D.W.; Kim, J.W.; Kim, B.; Kim, S.G. FXR Inhibits Endoplasmic Reticulum Stress-Induced NLRP3 Inflammasome in Hepatocytes and Ameliorates Liver Injury. Cell Rep. 2018, 24, 2985–2999. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, X.; Qiu, M.; Lv, S.; Liu, H. Hydrogen Sulfide Plays an Important Protective Role through Influencing Endoplasmic Reticulum Stress in Diseases. Int. J. Biol. Sci. 2020, 16, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, X.; Qiu, M.; Lv, S.; Zheng, H.; Niu, B.; Liu, H. Hydrogen Sulfide Plays an Important Role by Influencing NLRP3 inflammasome. Int. J. Biol. Sci. 2020, 16, 2752–2760. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| The Type of Pathological Processes | The Role of ER Stress and the NLRP3 Inflammasome | Experimental Model | Reference |
|---|---|---|---|
| Nonalcoholic hepatitis | ER stress promoted the NLRP3 inflammasome and subsequent pyroptosis and apoptosis to promote nonalcoholic hepatitis | Mouse/mouse primary hepatocyte model of nonalcoholic hepatitis | [46] |
| Nonalcoholic fatty liver disease (NAFLD) | BI-1 improved NAFLD through inhibition of ER stress-induced and IRE1a-dependent NLRP3 inflammasome activation | Mouse/mouse hepatocyte model of NAFLD | [50] |
| NAFLD | Rg1 improved NAFLD through the inhibition of ER stress and NLRP3 inflammasome activation | Mouse model of NAFLD | [54] |
| NAFLD | 4-AAQB ameliorated NAFLD through the inhibition of ERS/NLRP3 inflammasome by activating the SIRT1-Nrf2 pathway | Male C57BL/6J mouse model of NAFLD | [56] |
| Hepatic ischemia–reperfusion(HIRI) | ORY ameliorated HIRI through the inhibition of the NLRP3 inflammasome and ER stress | C57BL/6 mouse model of HIRI | [75] |
| Hepatotoxicity | Allicin improved AA-induced hepatotoxicity through ER stress inhibition of NLRP3 inflammasome via the MAPK and NF-κB signaling pathways | Sprague Dawley rats/Kupffer cell model of hepatotoxicity | [86] |
| Hepatotoxicity | BA improved PA-induced cytotoxicity of AML-12 cells through the inhibition of ER stress via the TXNIP/NLRP3 pathway, through the AMPK pathway | AML-12 cell model of hepatotoxicity | [95] |
| Liver injury | FXR improved liver injury through inhibition of ER stress-induced NLRP3 inflammasome via the PERK–CHOP signaling pathway | C57BL/6J mouse/AML-12 cell model of liver injury | [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Huang, H.; Fu, X.; Chen, C.; Liu, H.; Wang, H.; Wu, D. The Role of Endoplasmic Reticulum Stress and NLRP3 Inflammasome in Liver Disorders. Int. J. Mol. Sci. 2022, 23, 3528. https://doi.org/10.3390/ijms23073528
Lu X, Huang H, Fu X, Chen C, Liu H, Wang H, Wu D. The Role of Endoplasmic Reticulum Stress and NLRP3 Inflammasome in Liver Disorders. International Journal of Molecular Sciences. 2022; 23(7):3528. https://doi.org/10.3390/ijms23073528
Chicago/Turabian StyleLu, Xueqin, Haitao Huang, Xiaodi Fu, Chaoran Chen, Huiyang Liu, Honggang Wang, and Dongdong Wu. 2022. "The Role of Endoplasmic Reticulum Stress and NLRP3 Inflammasome in Liver Disorders" International Journal of Molecular Sciences 23, no. 7: 3528. https://doi.org/10.3390/ijms23073528