
Design and Experimental Evaluation of a Peptide Antagonist against Amyloid β(1–42) Interactions with Calmodulin and Calbindin-D28k

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Interaction between Aβ(1–42) HiLyteTM-Fluor555 and Calmodulin (CaM)
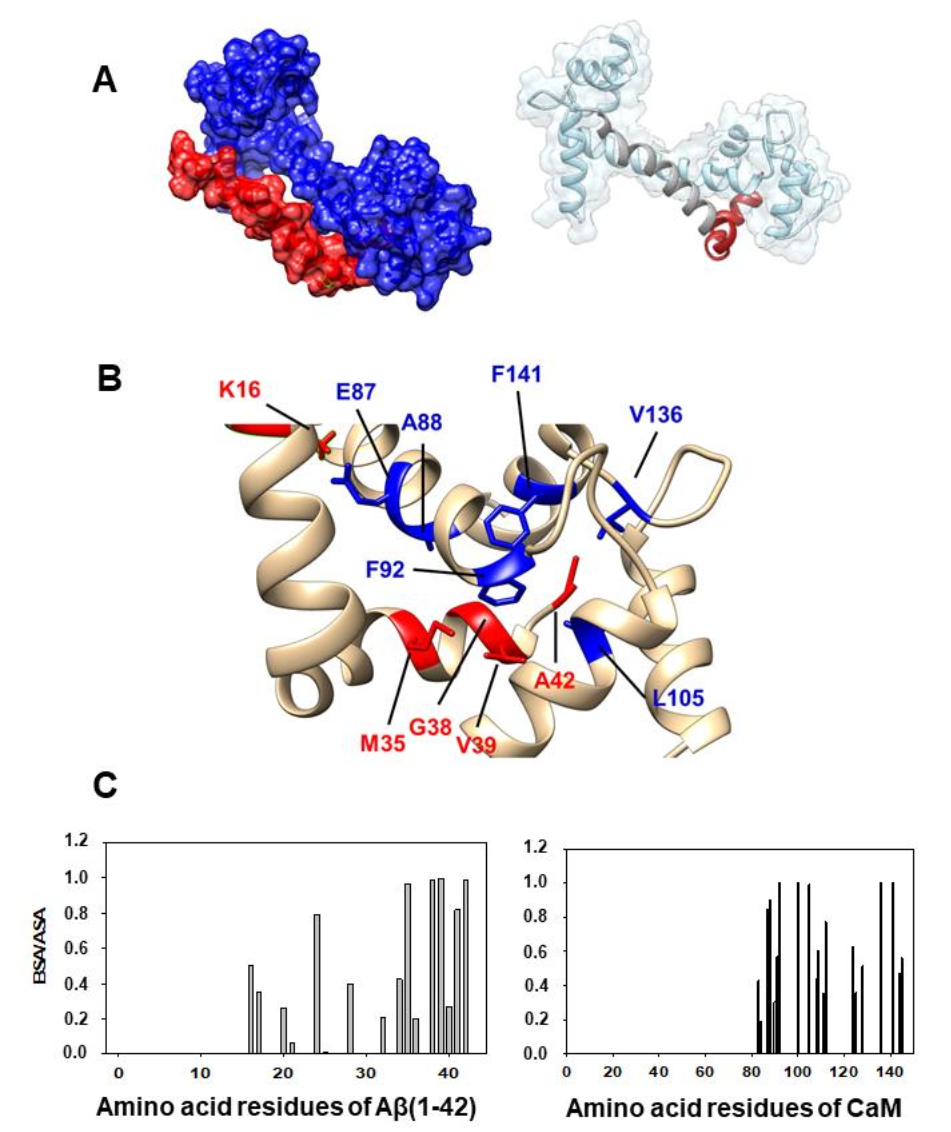
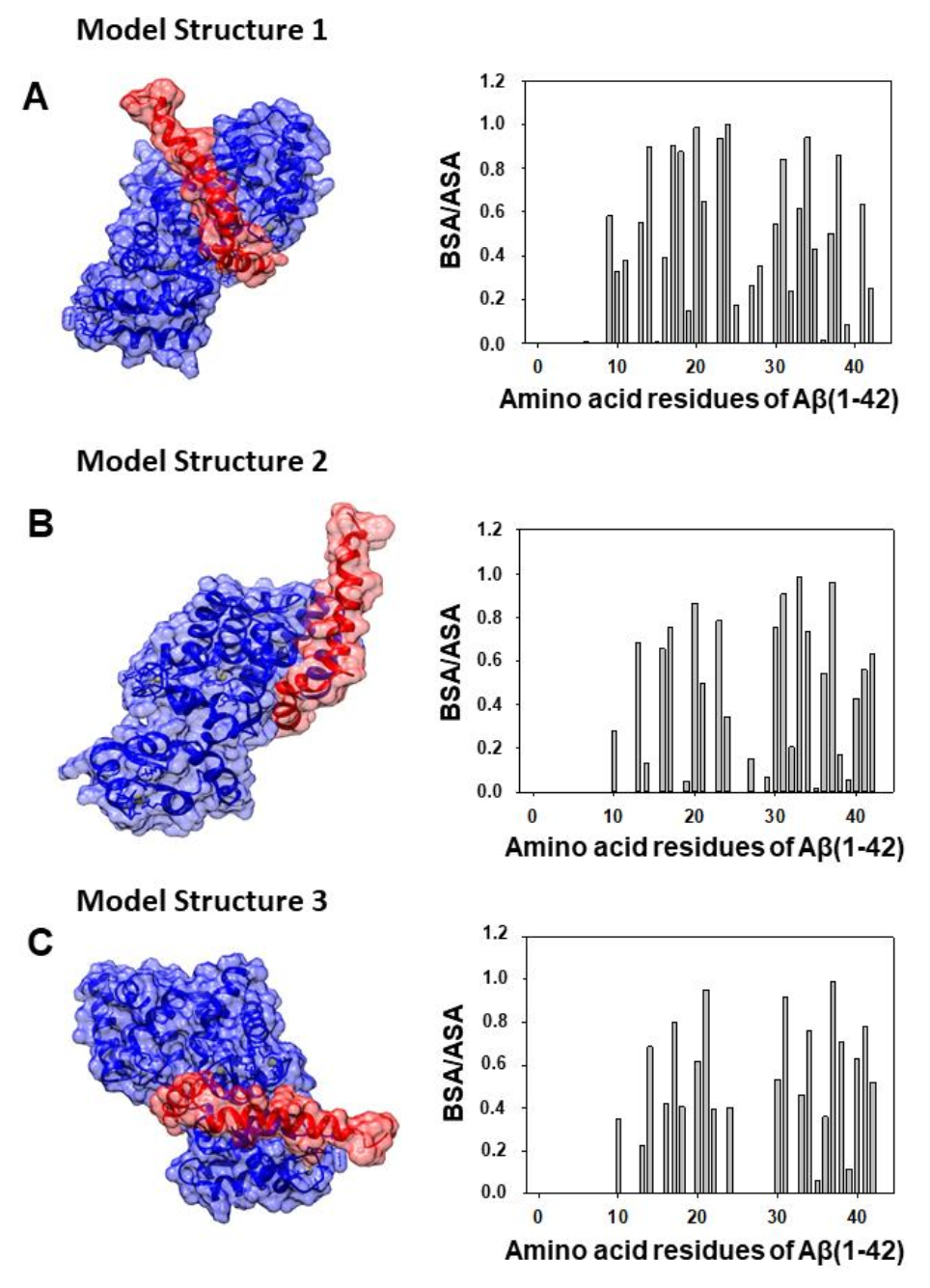
2.2. Docking between Aβ(1–42) and CaM and the Criteria to Select the Most Probable in Silico Structural Models of the Aβ(1–42):CaM Complex
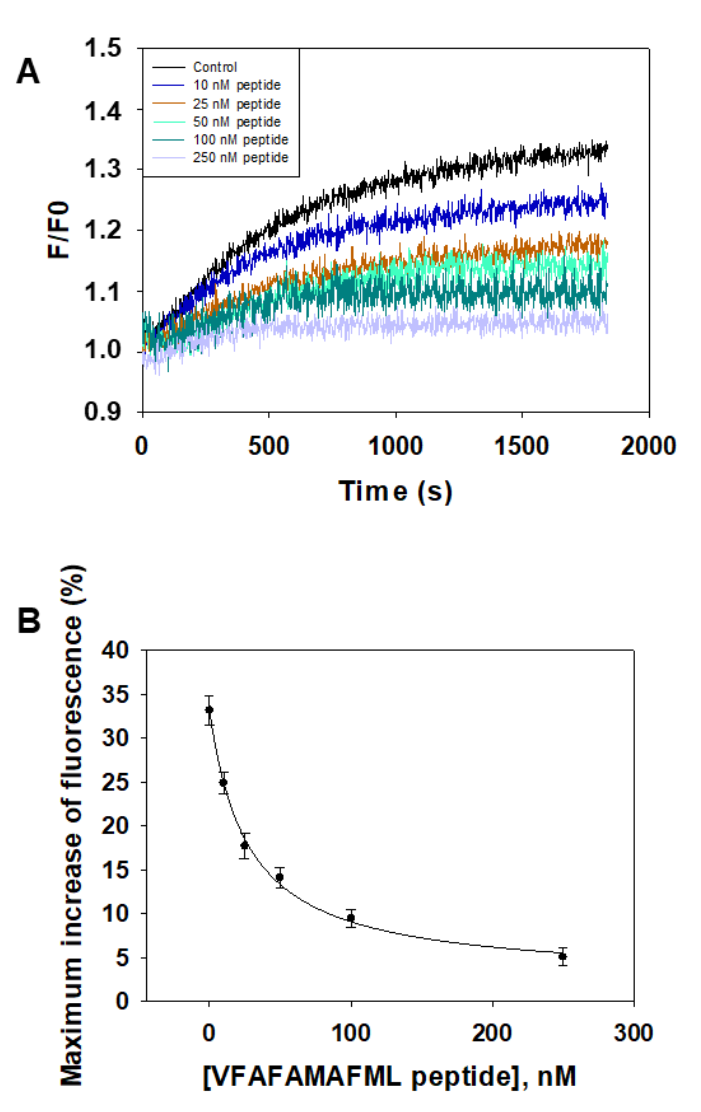
2.3. Design of an Antagonist Peptide for the Formation of the Aβ(1–42):CaM Complex and Its Experimental Evaluation
2.4. Interaction between Aβ(1–42) HiLyteTM-Fluor555 and Calbindin-D28k
2.5. Docking between Aβ(1–42) and Calbindin-D28k Gives Support to the Formation of Aβ(1–42):Calbindin-D28k Complexes
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Aβ(1–42) Solutions and Aggregation State
4.3. Expression and Purification of Calbindin-D28k
4.4. Measurements of Aβ(1–42) HiLyteTM-Fluor555 Interaction with CaM and Calbindin-D28k
4.5. In silico Docking Experiments
4.6. Experimental Evaluation of the Efficiency of the Designed Peptide to Antagonize the Interaction between Aβ(1–42) HiLyteTM-Fluor555 and CaM or Calbindin-D28k
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid β |
| AD | Alzheimer’s disease |
| BSA/ASA | buried surface area/accessible surface area ratio |
| CaM | calmodulin |
| DMSO | dimethylsulfoxide |
| Hepes | N-[2-hydroxyethyl] piperazine-N′-[2-ethanesulfonic acid] |
| IC50 | concentration producing 50% of the maximum effect |
| IPTG | isopropyl-β-d-thiogalactoside |
| LB | lysogeny broth |
| Qmax | maximum extinction of fluorescence |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| S.E. | standard error |
| Tris | tris-(hydroxymethyl) aminomethane |
References
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.-M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-β peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, R.P.; Tepper, K.; Rönicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fändrich, M. Mechanism of amyloid plaque formation suggests an intracellular basis of Aβ pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Kedia, N.; Illes-Toth, E.; Haralampiev, I.; Prisner, S.; Herrmann, A.; Wanker, E.E.; Bieschke, J. Amyloid-β(1–42) Aggregation Initiates Its Cellular Uptake and Cytotoxicity. J. Biol. Chem. 2016, 291, 19590–19606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirths, O.; Multhaup, G.; Czech, C.; Blanchard, V.; Moussaoui, S.; Tremp, G.; Pradier, L.; Beyreuther, K.; Bayer, T.A. Intraneuronal Aβ accumulation precedes plaque formation in β-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 2001, 306, 116–120. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic arcAβ mice. Neurobiol. Aging 2007, 28, 1297–1306. [Google Scholar] [CrossRef]
- Zhang, Y.; McLaughlin, R.; Goodyer, C.G.; Leblanc, A.C. Selective cytotoxicity of intracellular amyloid β peptide1–42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 2002, 156, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.-Y.; Hyman, B.T.; Bacskai, B.J. Aβ Plaques Lead to Aberrant Regulation of Calcium Homeostasis In Vivo Resulting in Structural and Functional Disruption of Neuronal Networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.R.; Lyckman, A.; Oddo, S.; LaFerla, F.M.; Querfurth, H.W.; Shtifman, A. Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J. Neurochem. 2008, 105, 262–271. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium Signalling and Alzheimer’s Disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef]
- Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. The Relevance of Amyloid β-Calmodulin Complexation in Neurons and Brain Degeneration in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4976. [Google Scholar] [CrossRef]
- Biber, A.; Schmid, G.; Hempel, K. Calmodulin content in specific brain areas. Exp. Brain Res. 1984, 56, 323–326. [Google Scholar] [CrossRef]
- Xia, Z.; Storm, D.R. The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 2005, 6, 267–276. [Google Scholar] [CrossRef]
- Corbacho, I.; Berrocal, M.; Török, K.; Mata, A.M.; Gutierrez-Merino, C. High affinity binding of amyloid β-peptide to calmodulin: Structural and functional implications. Biochem. Biophys. Res. Commun. 2017, 486, 992–997. [Google Scholar] [CrossRef] [Green Version]
- Poejo, J.; Salazar, J.; Mata, A.; Gutierrez-Merino, C. Binding of Amyloid β(1–42)-Calmodulin Complexes to Plasma Membrane Lipid Rafts in Cerebellar Granule Neurons Alters Resting Cytosolic Calcium Homeostasis. Int. J. Mol. Sci. 2021, 22, 1984. [Google Scholar] [CrossRef]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nat. Cell Biol. 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [Green Version]
- Dunning, C.J.; McGauran, G.; Willén, K.; Gouras, G.K.; O’Connell, D.J.; Linse, S. Direct High Affinity Interaction between Aβ42 and GSK3α Stimulates Hyperphosphorylation of Tau. A New Molecular Link in Alzheimer’s Disease? ACS Chem. Neurosci. 2015, 7, 161–170. [Google Scholar] [CrossRef] [Green Version]
- O’Day, D.H. Calmodulin Binding Proteins and Alzheimer’s Disease: Biomarkers, Regulatory Enzymes and Receptors That Are Regulated by Calmodulin. Int. J. Mol. Sci. 2020, 21, 7344. [Google Scholar] [CrossRef]
- Christakos, S.; Gabrielides, C.; Rhoten, W.B. Vitamin D-dependent calcium binding proteins: Chemistry, distribution, functional considerations and molecular biology. Endocr. Rev. 1989, 10, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Pasti, L.; Carmignoto, G.; Pozzan, T.; Battini, R.; Ferrari, S.; Lally, G.; Emson, P.C. Cellular calcium handling in brain slices from calbindin D28k-deficient mice. Neuroreport 1999, 10, 2367–2372. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Jones, B.; Kekonius, L.; Chin, J.; Yu, G.-Q.; Raber, J.; Masliah, E.; Mucke, L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease related cognitive deficits. Proc. Natl. Acad. Sci. USA 2003, 100, 9572–9577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, J.R.; Radenahmad, N.; Wilcock, G.K.; Neal, J.W.; Pearson, R.C. Accumulation of calbindin in cortical pyramidal cells with ageing; a putative protective mechanism which fails in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 339–342. [Google Scholar] [CrossRef]
- Guo, Q.; Christakos, S.; Robinson, N.; Mattson, M.P. Calbindin D28k blocks the proapoptotic actions of mutant presenilin 1: Reduced oxidative stress and preserved mitochondrial function. Proc. Natl. Acad. Sci. USA 1998, 95, 3227–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernyj, R.P.; Mattson, M.P.; Christakos, S. Expression of calbindin-D28k in C6 glial cells stabilizes intracellular calcium levels and protects against apoptosis induced by calcium ionophore and amyloid beta-peptide. Brain Res. Mol. Brain Res. 1999, 64, 69–79. [Google Scholar] [CrossRef]
- Kook, S.-Y.; Jeong, H.; Kang, M.J.; Park, R.; Shin, H.J.; Han, S.-H.; Son, S.M.; Song, H.; Baik, S.H.; Moon, M.; et al. Crucial role of calbindin-D28k in the pathogenesis of Alzheimer’s disease mouse model. Cell Death Differ. 2014, 21, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, V.V.; Santos, D.B.; Lach, G.; Rodrigues, A.L.; Farina, M.; De Lima, T.C.; Prediger, R.D. Neuropeptide Y (NPY) prevents depressive-like behavior, spatial memory deficits and oxidative stress following amyloid-β [Aβ(1-40)] administration in mice. Behav. Brain Res. 2013, 244, 107–115. [Google Scholar] [CrossRef]
- Kang, S.; Moon, N.R.; Kim, D.S.; Kim, S.H.; Park, S. Central acylated ghrelin improves memory function and hippocampal AMPK activation and partly reverses the impairment of energy and glucose metabolism in rats infused with β-amyloid. Peptides 2015, 71, 84–93. [Google Scholar] [CrossRef]
- Santos, V.V.; Stark, R.; Rial, D.; Silva, H.B.; Bayliss, J.A.; Lemus, M.B.; Davies, J.S.; Cunha, R.A.; Prediger, R.D.; Andrews, Z.B. Acyl ghrelin improves cognition, synaptic plasticity deficits and neuroinflammation following amyloid β (Aβ1-40) administration in mice. J. Neuroendocrinol. 2017, 29, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.; Gomes, S.; Costa, R.O.; Otvos, L.; Oliveira, C.R.; Resende, R.; Pereira, C.M.F. Leptin and ghrelin prevent hippocampal dysfunction induced by Aβ oligomers. Neuroscience 2013, 241, 41–51. [Google Scholar] [CrossRef]
- Gomes, S.; Martins, I.; Fonseca, A.C.; Oliveira, C.R.; Resende, R.; Pereira, C.M. Protective effect of leptin and ghrelin against toxicity induced by amyloid-β oligomers in a hypothalamic cell line. J. Neuroendocrinol. 2014, 26, 176–185. [Google Scholar] [CrossRef]
- Baimbridge, K.G.; Celio, M.R.; Rogers, J.H. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992, 15, 303–308. [Google Scholar] [CrossRef]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid beta-peptide (1–42) in an apolar microenvironment: Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef]
- Tomaselli, S.; Esposito, V.; Vangone, P.; van Nuland, N.A.J.; Bonvin, A.M.J.J.; Guerrini, R.; Tancredi, T.; Temussi, P.A.; Picone, D. The α-to-β Conformational Transition of Alzheimer’s Aβ-(1–42) Peptide in Aqueous Media is Reversible: A Step by Step Conformational Analysis Suggests the Location of β Conformation Seeding. ChemBioChem 2006, 7, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.; Siegel, G.; Wayne Albers, R.; Price, D.L. Basic Neurochemistry: Principles of Molecular, Cellular, and Medical Neurobiology, 8th ed.; Academic Press/Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Bahr, B.A.; Hoffman, K.B.; Yang, A.J.; Hess, U.S.; Glabe, C.G.; Lynch, G. Amyloid β protein is internalized selectively by hippocampal field CA1 and causes neurons to accumulate amyloidogenic carboxyterminal fragments of the amyloid precursor protein. J. Comp. Neurol. 1998, 397, 139–147. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Umahara, T.; Hanyu, H.; Iwamoto, T.; Matsuoka, M. Calmodulin-like skin protein is downregulated in human cerebrospinal fluids of Alzheimer’s disease patients with apolipoprotein E4; a pilot study using postmortem samples. Neurol. Res. 2017, 39, 767–772. [Google Scholar] [CrossRef]
- Kusakari, S.; Nawa, M.; Sudo, K.; Matsuoka, M. Calmodulin-like skin protein protects against spatial learning impairment in a mouse model of Alzheimer disease. J. Neurochem. 2018, 144, 218–233. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Nawa, M.; Kurita, M.; Tokizawa, M.; Iwamatsu, A.; Matsuoka, M. Secreted calmodulin-like skin protein inhibits neuronal death in cell-based Alzheimer’s disease models via the heterotrimeric Humanin receptor. Cell Death Dis. 2013, 4, e555. [Google Scholar] [CrossRef] [Green Version]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid beta peptide 25-35. Curr. Protein Pept. Sci. 2010, 11, 54–67. [Google Scholar] [CrossRef]
- Pike, C.J.; Walencewicz-Wasserman, A.J.; Kosmoski, J.; Cribbs, D.H.; Glabe, C.G.; Cotman, C.W. Structure-activity analyses of beta-amyloid peptides: Contributions of the beta 25–35 region to aggregation and neurotoxicity. J. Neurochem. 1995, 64, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Clementi, M.E.; Marini, S.; Colette, M.; Orsini, F.; Giardina, B.; Misiti, F. Aβ(31–35) and Aβ(25–35) fragments of amyloid beta-protein induce cellular death through apoptotic signals: Role of the redox state of methionine-35. FEBS Lett. 2005, 579, 2913–2918. [Google Scholar] [CrossRef] [PubMed]
- Frozza, R.L.; Horn, A.P.; Hoppe, J.B.; Simão, F.; Gerhardt, D.; Comiran, R.A.; Salbego, C.G. A comparative study of beta-amyloid peptides Abeta1–42 and Abeta25–35 toxicity in organotypic hippocampal slice cultures. Neurochem. Res. 2009, 34, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Bascuñán, D.; Opazo, C.; Aguayo, L.G. Differential membrane toxicity of amyloid-β fragments by pore forming mechanisms. J. Alzheimers Dis. 2016, 51, 689–699. [Google Scholar] [CrossRef]
- D’Ezio, V.; Colasanti, M.; Persichini, T. Amyloid-β 25-35 Induces Neurotoxicity through the Up-Regulation of Astrocytic System Xc¯. Antioxidants 2021, 10, 1685. [Google Scholar] [CrossRef]
- Guo, J.-P.; Arai, T.; Miklossy, J.; McGeer, P.L. Aβ and tau form soluble complexes that may promote self-aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.B.; Crews, L.; Rockenstein, E.; Adame, A.; Mante, M.; Hersh, L.B.; Gage, F.H.; Spencer, B.; Potkar, R.; Marr, R.A.; et al. Neuropeptide Y Fragments Derived from Neprilysin Processing Are Neuroprotective in a Transgenic Model of Alzheimer’s Disease. J. Neurosci. 2009, 29, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-Y.; Du, Y.-F.; Chen, L. Neuropeptides Exert Neuroprotective Effects in Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 11, 493. [Google Scholar] [CrossRef] [Green Version]
- Gahete, M.D.; Rubio, A.; Córdoba-Chacón, J.; Gracia-Navarro, F.; Kineman, R.D.; Avila, J.; Luque, R.M.; Castaño, J.P. Expression of the ghrelin and neurotensin systems is altered in the temporal lobe of Alzheimer’s disease patients. J. Alzheimers Dis. 2010, 22, 819–828. [Google Scholar] [CrossRef]
- Shibata, N.; Ohnuma, T.; Kuerban, B.; Komatsu, M.; Arai, H. Genetic association between ghrelin polymorphisms and Alzheimer’s disease in a Japanese population. Dement. Geriatr. Cogn. Disord. 2011, 32, 178–181. [Google Scholar] [CrossRef]
- Noble, J.W.; Almalki, R.; Roe, S.M.; Wagner, A.; Duman, R.; Atack, J.R. The X-ray structure of human calbindin-D28K: An improved model. Acta Cryst. 2018, D74, 1008–1014. [Google Scholar] [CrossRef]
- Tao, L.; Murphy, M.E.; English, A.M. S-nitrosation of Ca(2+)-loaded and Ca(2+)-free recombinant calbindin D(28K) from human brain. Biochemistry 2002, 41, 6185–6192. [Google Scholar] [CrossRef]
- Bauer, M.C.; Nilsson, H.; Thulin, E.; Frohm, B.; Malm, J.; Linse, S. Zn2+ binding to human calbindin D(28k) and the role of histidine residues. Protein Sci. 2008, 17, 760–767. [Google Scholar] [CrossRef] [Green Version]
- Blaszczyk, M.; Kurcinski, M.; Kouza, M.; Wieteska, L.; Debinski, A.; Kolinski, A.; Kmiecik, S. Modeling of protein-peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods 2016, 93, 72–83. [Google Scholar] [CrossRef]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aβ(1–42) Amino Acid Residues | CaM Amino Acid Residues | |
|---|---|---|
| Model structure 1 | Gly37 (1.0), Gly38 (1.0), Val36 (0.98), Phe20 (0.97), Val40 (0.89), Leu34 (0.83), Val39 (0.82), Met35 (0.77), Lys28 (0.72), Val24 (0.66) | Ile27 (1.0), Leu32 (1.0), Ile52 (1.0), Ile63 (1.0), Phe68 (1.0), Met71 (1.0), Phe19 (0.99), Phe89 (0.99), Gly98 (0.98), Val55 (0.89), Met51 (0.88), Arg86 (0.85), Met36 (0.76), Met72 (0.74), Lys75 (0.73), Thr79 (0.70), Gly96 (0.68), Asn97 (0.64) |
| Model structure 2 | Val39 (0.99), Ala42 (0.99), Gly38 (0.98), Met35 (0.96), Ile41 (0.82), Val24 (0.79), Lys16 (0.5) | Val136 (1.0), Phe141 (1.0), Phe92 (0.997), Leu105 (0.99), Ala88 (0.90), Glu87 (0.85), Leu112 (0.77), Met124 (0.63), Met109 (0.61), Val91 (0.57), Met145 (0.56), Ala128 (0.52) |
| Aβ(1–42) Amino Acid Residues | Calbindin-D28k Amino Acid Residues | |
|---|---|---|
| Model structure 1 | Val24 (1), Phe20 (0.98), Leu34 (0.94), Asp23 (0.93), Leu17 (0.91), His14 (0.90), Val18 (0.88), Gly38 (0.86), Ile31 (0.84), Ala21 (0.65), Ile41 (0.63), Gly33 (0.62), Ala30 (0.55), Gly37 (0.52) | Val173 (0.96), Ile73 (0.94), Val181 (0.88), Leu179 (0.86), Lys72 (0.85), Glu177 (0.85), His80 (0.84), Ser28 (0.83), Glu57 (0.82), Phe61 (0.81), Pro172 (0.68), Tyr30 (0.67), Ser156 (0.61) |
| Model structure 2 | Gly33 (0.98), Gly37 (0.96), Ile31 (0.91), Phe20 (0.86), Asp23 (0.78), Leu17 (0.75), Ala30 (0.75), Leu34 (0.74), His13 (0.68), Lys16 (0.66), Ala42 (0.63), Ile41 (0.55), Val36 (0.54) | His80 (0.95), Arg47 (0.95), Pro172 (0.92), Leu89 (0.82), Glu57 (0.78), Arg93 (0.77), Leu82 (0.76), Lys98 (0.74), Ser55 (0.74), Glu77 (0.73), Pro83 (0.72), Leu52 (0.71), Val81 (0.63) |
| Model structure 3 | Gly37 (0.99), Ala21 (0.95), Ile31 (0.92), Leu17 (0.80), Ile41 (0.78), Leu34 (0.76), Gly38 (0.71), His14 (0.69), Val40 (0.63), Phe20 (0.62), Ala30 (0.53), Ala42 (0.52), | Gly188 (0.99), Ser28 (0.95), Asn157 (0.87), Ala258 (0.84), Glu166 (0.83), Phe177 (0.80), Asn192 (0.72), Cys257 (0.70), Arg249 (0.70), Ser156 (0.67), Phe191 (0.67), Thr250 (0.63), Gln182 (0.63) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salazar, J.; Poejo, J.; Mata, A.M.; Samhan-Arias, A.K.; Gutierrez-Merino, C. Design and Experimental Evaluation of a Peptide Antagonist against Amyloid β(1–42) Interactions with Calmodulin and Calbindin-D28k. Int. J. Mol. Sci. 2022, 23, 2289. https://doi.org/10.3390/ijms23042289
Salazar J, Poejo J, Mata AM, Samhan-Arias AK, Gutierrez-Merino C. Design and Experimental Evaluation of a Peptide Antagonist against Amyloid β(1–42) Interactions with Calmodulin and Calbindin-D28k. International Journal of Molecular Sciences. 2022; 23(4):2289. https://doi.org/10.3390/ijms23042289
Chicago/Turabian StyleSalazar, Jairo, Joana Poejo, Ana M. Mata, Alejandro K. Samhan-Arias, and Carlos Gutierrez-Merino. 2022. "Design and Experimental Evaluation of a Peptide Antagonist against Amyloid β(1–42) Interactions with Calmodulin and Calbindin-D28k" International Journal of Molecular Sciences 23, no. 4: 2289. https://doi.org/10.3390/ijms23042289