
Mechanisms Underlying the Expansion and Functional Maturation of β-Cells in Newborns: Impact of the Nutritional Environment


Abstract
:1. Introduction
2. Literature Search Strategy
3. Insights into the Intracellular Mechanisms Driving Early Postnatal β-Cell Mass Expansion and Maturation
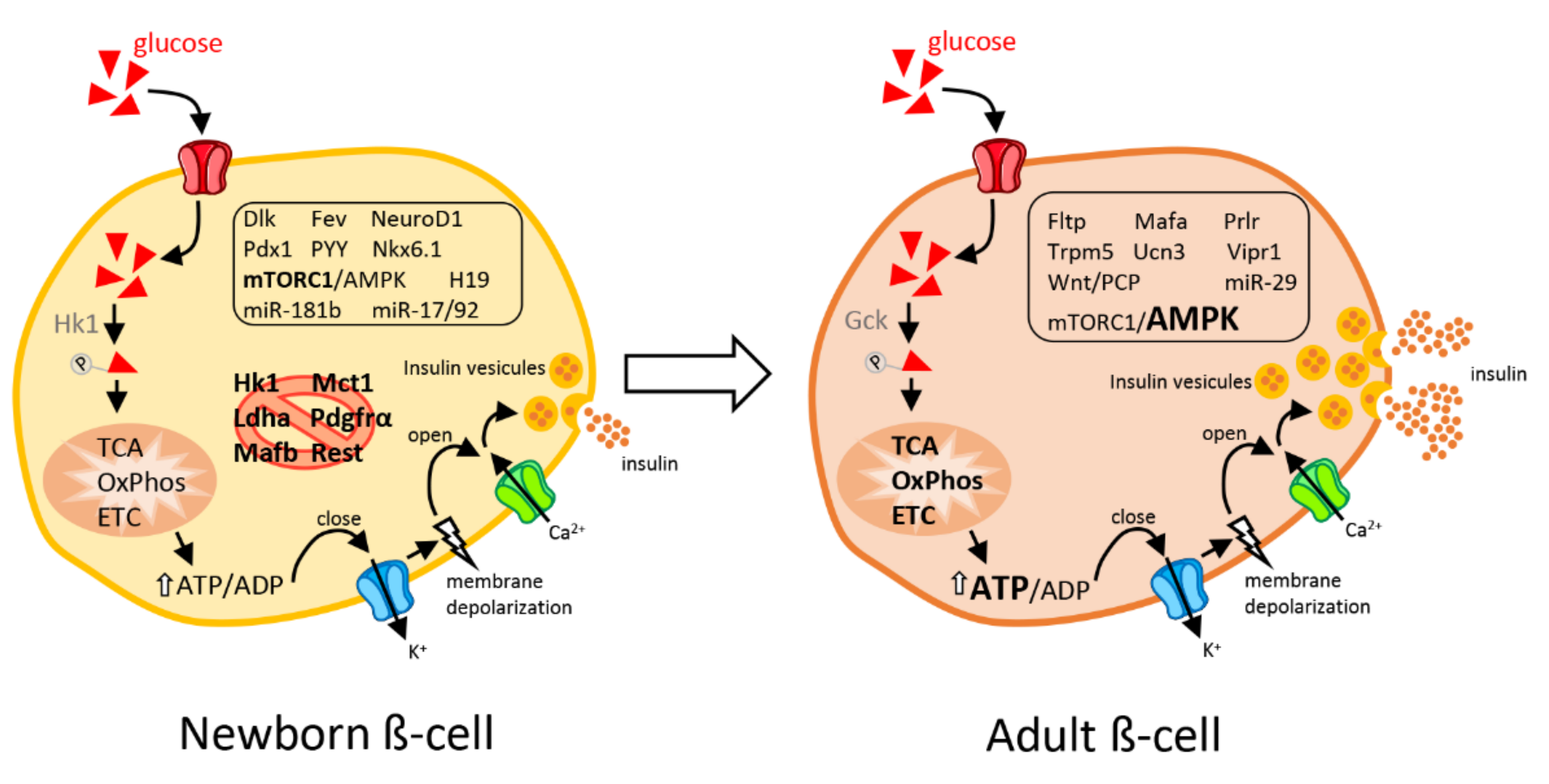
3.1. β-Cell Heterogeneity during Postnatal Development and “Immaturity Signature”
3.2. New Insights into the Molecular Mechanisms of β-Cell Replication in Newborns
3.3. Signaling Pathways Driving the Acquisition of Functional β-Cell Features
3.4. Environmental Drivers of the Transition from Immature to Mature Postnatal β-Cell Function
4. Inappropriate Fetal and Postnatal Environment Induces Persistent Changes That Increase Type 2 Diabetes Risk
4.1. In Utero and Postnatal “Obesogenic” Environment has a Deleterious Impact on β-Cells of the Progeny
4.2. Effects of Maternal Undernutrition on β-Cells of the Progeny
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ABCC8 | ATP binding cassette subfamily C member 8 |
| ADP | adenosine diphosphate |
| AKT | a serine/threonine protein kinase |
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| Atp5f1 | ATP synthase peripheral stalk-membrane subunit B |
| BMP4 | bone morphogenetic protein 4 |
| Ca2+ | calcium ions |
| Cacna1c/d | calcium voltage-gated channel subunit alpha1 C/D |
| CASR | calcium-sensing receptor |
| CCL2 | C-C Motif Chemokine Ligand 2 |
| Ccnc | cyclin C |
| Ccnd1/2 | cyclin D1/2 |
| CDK1 | cyclin-dependent kinase 1 |
| Cdt1 | chromatin licensing and DNA replication factor 1 |
| CENPA | centromere protein A |
| c-Myc | myelocytomatosis viral proto-oncogene, BHLH transcription factor |
| Deptor | DEP domain containing MTOR interacting protein |
| Dlk | dual leucine zipper-bearing kinase |
| DNA | deoxyribonucleic acid |
| DNMT3A | DNA methyltransferase 3 alpha |
| ETC | electron transport chain |
| Fabp5 | fatty acid-binding protein 5 |
| FACS | fluorescence-activated cell sorting |
| Fev | fifth ewing variant |
| Fltp | flattop |
| FOXM1 | forkhead box protein M1 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| Gas6 | growth arrest specific protein 6 |
| Gck | glucokinase |
| Glp1 | glucagon-like peptide 1 |
| GSIS | glucose-stimulated insulin secretion |
| H19 | H19 imprinted maternally expressed transcript |
| H3K4 | histone H3 lysine K4 |
| HFD | high-fat diet |
| Hk | hexokinase 1 |
| Hnf1a/4a | hepatocyte nuclear factor-1/4 alpha |
| IL1beta | interleukin 1 beta |
| IL-1Ra | interleukin 1 receptor antagonist |
| Il13ra2 | interleukin 13 receptor subunit alpha 2 |
| iPSC | induced pluripotent stem cells |
| Isl1 | insulin gene enhancer protein |
| IUGR | intrauterine growth retardation |
| Jak | janus kinase |
| Jnk3 | c-Jun N-terminal kinase 3 |
| K+ | a positively charged potassium ion |
| Ldha | lactate dehydrogenase A |
| LP | low protein |
| Maea | macrophage erythroblast attacher, E3 ubiquitin ligase |
| Mafa | basic leucine zipper (bZIP) transcription factor A |
| Mafb | basic leucine zipper (bZIP) transcription factor B |
| Mcm3/4/6/7/10 | minichromosome maintenance complex component 3/4/6/7/10 |
| Mct1 | monocarboxylate transporter 1 |
| Mettl3/14 | RNA methyltransferase-like 3/14 |
| MIN6 | mouse insulinoma cell line |
| mTORC1/2 | mammalian target of rapamycin complex 1/2 |
| NADH | nicotinamide adenine dinucleotide |
| NeuroD1 | neuronal differentiation 1 |
| Nkx6.1 | NK6 homeobox 1 |
| Orc5/6 | origin recognition complex subunit 5/6 |
| OxPhos | oxidative phosphorylation |
| PCP | planar cell polarity |
| Pdgfrα | platelet derived growth factor receptor alpha |
| Pdk1 | phosphoinositide-dependent kinase-1 |
| Pdx1 | pancreatic and duodenal homeobox 1 |
| PI3K | phosphoinositide 3-kinase |
| PLA2 | phospholipase A2 |
| Prim1 | DNA Primase Subunit 1 |
| Prlr | prolactin receptor |
| PYY | peptide YY |
| Rest | RE1 silencing transcription factor |
| Rfx6 | regulatory factor X6 |
| RNA | ribonucleic acid |
| Rps6/14 | ribosomal protein S6/14 |
| SIX3 | Sine Oculis homeobox homolog 3 |
| Slc2a2 | solute carrier family 2 member 2 |
| SMAD2/3 | TGFB signaling protein, mothers against decapentaplegic homolog 2/3 |
| Sod2 | superoxide dismutase 2 |
| Stat3 | signal transducer and activator of transcription 3 |
| T2DM | type 2 diabetes mellitus |
| TCA | tricarboxylic acid cycle |
| Tfam | transcription factor A, mitochondrial |
| TGFBR | transforming growth factor beta receptor |
| TH | thyroid hormone |
| TK | transketolase |
| TOP2A | DNA Topoisomerase II Alpha |
| tRF | tRNA-derived fragment |
| tRNA | transfer RNA |
| Trpm5 | transient receptor potential cation channel subfamily M member 5 |
| Ucn3 | urocortin 3 |
| Vipr1 | vasoactive intestinal polypeptide receptor 1 |
| Wnt | wingless/integrated |
References
- Bonner-Weir, S.; Aguayo-Mazzucato, C.; Weir, G.C. Dynamic development of the pancreas from birth to adulthood. Upsala J. Med. Sci. 2016, 121, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Mezza, T.; Kulkarni, R.N. The regulation of pre- and post-maturational plasticity of mammalian islet cell mass. Diabetologia 2014, 57, 1291–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.S.E.; Hebrok, M. All mixed up: Defining roles for beta-cell subtypes in mature islets. Genes Dev. 2017, 31, 228–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, A.M.; Gannon, M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J. Mol. Endocrinol. 2007, 38, 193–206. [Google Scholar] [CrossRef]
- Patel, M.S.; Srinivasan, M. Metabolic Programming Due to Alterations in Nutrition in the Immediate Postnatal Period. J. Nutr. 2010, 140, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Pullen, T.J.; Hodson, D.J.; Martinez-Sanchez, A. Pancreatic beta-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 2015, 466, 203–218. [Google Scholar] [CrossRef] [Green Version]
- Guay, C.; Jacovetti, C.; Bayazit, M.B.; Brozzi, F.; Rodriguez-Trejo, A.; Wu, K.J.; Regazzi, R. Roles of Noncoding RNAs in Islet Biology. Compr. Physiol. 2020, 10, 893–932. [Google Scholar] [CrossRef]
- Avrahami, D.; Wang, Y.J.; Schug, J.; Feleke, E.; Gao, L.; Liu, C.Y.; Naji, A.; Glaser, B.; Kaestner, K.H.; Consortium, H. Single-cell transcriptomics of human islet ontogeny defines the molecular basis of beta-cell dedifferentiation in T2D. Mol. Metab. 2020, 42, 101057. [Google Scholar] [CrossRef]
- Arda, H.E.; Benitez, C.M.; Kim, S.K. Gene Regulatory Networks Governing Pancreas Development. Dev. Cell 2013, 25, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Martens, G.A.; Motte, E.; Kramer, G.; Stange, G.; Gaarn, L.W.; Hellemans, K.; Nielsen, J.H.; Aerts, J.M.; Ling, Z.; Pipeleers, D. Functional characteristics of neonatal rat beta cells with distinct markers. J. Mol. Endocrinol. 2014, 52, 11–28. [Google Scholar] [CrossRef]
- Stolovich-Rain, M.; Enk, J.; Vikesa, J.; Nielsen, F.C.; Saada, A.; Glaser, B.; Dor, Y. Weaning Triggers a Maturation Step of Pancreatic beta Cells. Dev. Cell 2015, 33, 238–239. [Google Scholar] [CrossRef] [Green Version]
- Puri, S.; Roy, N.; Russ, H.A.; Leonhardt, L.; French, E.K.; Roy, R.; Bengtsson, H.; Scott, D.K.; Stewart, A.F.; Hebrok, M. Replication confers beta cell immaturity. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-L.; Zhang, Y.-W.; Feng, Y.; Li, L.-C.; Yang, L.; Xu, C.-R. Deciphering Pancreatic Islet beta Cell and alpha Cell Maturation Pathways and Characteristic Features at the Single-Cell Level. Cell Metab. 2017, 25, 1194–1205.e4. [Google Scholar] [CrossRef] [Green Version]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef]
- Tritschler, S.; Theis, F.J.; Lickert, H.; Böttcher, A. Systematic single-cell analysis provides new insights into heterogeneity and plasticity of the pancreas. Mol. Metab. 2017, 6, 974–990. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, U.; Hudgens, C.W.; Wright, B.T.; Maulis, M.F.; Gannon, M. Differential regulation of embryonic and adult beta cell replication. Cell Cycle 2012, 11, 2431–2442. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Hang, Y.; Shostak, A.; Poffenberger, G.; Hart, N.; Prasad, N.; Phillips, N.; Levy, S.E.; Greiner, D.L.; Shultz, L.D.; et al. Age-dependent human beta cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J. Clin. Investig. 2017, 127, 3835–3844. [Google Scholar] [CrossRef]
- Sanchez-Parra, C.; Jacovetti, C.; Dumortier, O.; Lee, K.; Peyot, M.-L.; Guay, C.; Prentki, M.; Laybutt, D.R.; Van Obberghen, E.; Regazzi, R. Contribution of the Long Noncoding RNA H19 to beta-Cell Mass Expansion in Neonatal and Adult Rodents. Diabetes 2018, 67, 2254–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatrai, S.; Elghazi, L.; Balcazar, N.; Cras-Méneur, C.; Krits, I.; Kiyokawa, H.; Bernal-Mizrachi, E. Akt Induces beta-Cell Proliferation by Regulating Cyclin D1, Cyclin D2, and p21 Levels and Cyclin-Dependent Kinase-4 Activity. Diabetes 2006, 55, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Arda, H.E.; Li, L.; Tsai, J.; Torre, E.A.; Rosli, Y.; Peiris, H.; Spitale, R.C.; Dai, C.H.; Gu, X.Y.; Qu, K.; et al. Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human beta Cell Function. Cell Metab. 2016, 23, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Tenenbaum, M.; Plaisance, V.; Boutry, R.; Pawlowski, V.; Jacovetti, C.; Sanchez-Parra, C.; Ezanno, H.; Bourry, J.; Beeler, N.; Pasquetti, G.; et al. The Map3k12 (Dlk)/JNK3 signaling pathway is required for pancreatic beta-cell proliferation during postnatal development. Cell. Mol. Life Sci. 2021, 78, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Hanley, S.C.; Austin, E.; Assouline-Thomas, B.; Kapeluto, J.; Blaichman, J.; Moosavi, M.; Petropavlovskaia, M.; Rosenberg, L. beta-Cell Mass Dynamics and Islet Cell Plasticity in Human Type 2 Diabetes. Endocrinology 2010, 151, 1462–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnemann, A.K.; Baan, M.; Davis, D.B. Pancreatic beta-Cell Proliferation in Obesity. Adv. Nutr. Int. Rev. J. 2014, 5, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jermendy, A.; Toschi, E.; Aye, T.; Koh, A.; Aguayo-Mazzucato, C.; Sharma, A.; Weir, G.C.; Sgroi, D.; Bonner-Weir, S. Rat neonatal beta cells lack the specialised metabolic phenotype of mature beta cells. Diabetologia 2011, 54, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, H.L.; Corbin, K.L.; D’Angelo, C.V.; Donovan, L.M.; Jahan, I.; Gu, G.Q.; Nunemaker, C.S. Postnatal maturation of calcium signaling in islets of Langerhans from neonatal mice. Cell Calcium 2021, 94, 102339. [Google Scholar] [CrossRef]
- Osipovich, A.B.; Dudek, K.D.; Greenfest-Allen, E.; Cartailler, J.-P.; Manduchi, E.; Case, L.P.; Choi, E.; Chapman, A.G.; Clayton, H.W.; Gu, G.Q.; et al. A developmental lineage-based gene co-expression network for mouse pancreatic beta-cells reveals a role for Zfp800 in pancreas development. Development 2021, 148, 1802–1812. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Koh, A.; El Khattabi, I.; Li, W.-C.; Toschi, E.; Jermendy, A.; Juhl, K.; Mao, K.; Weir, G.C.; Sharma, A.; et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 2011, 54, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Henaoui, I.S.; Jacovetti, C.; Mollet, I.G.; Guay, C.; Sobel, J.; Eliasson, L.; Regazzi, R. PIWI-interacting RNAs as novel regulators of pancreatic beta cell function. Diabetologia 2017, 60, 1977–1986. [Google Scholar] [CrossRef]
- Jacovetti, C.; Matkovich, S.J.; Rodríguez-Trejo, A.; Guay, C.; Regazzi, R. Postnatal beta-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nat. Commun. 2015, 6, 8084. [Google Scholar] [CrossRef] [Green Version]
- Bevacqua, R.J.; Lam, J.Y.; Peiris, H.; Whitener, R.L.; Kim, S.; Gu, X.Y.; Friedlander, M.S.H.; Kim, S.K. SIX2 and SIX3 coordinately regulate functional maturity and fate of human pancreatic beta cells. Genes Dev. 2021, 35, 234–249. [Google Scholar] [CrossRef]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Thorrez, L.; Laudadio, I.; Van Deun, K.; Quintens, R.; Hendrickx, N.; Granvik, M.; Lemaire, K.; Schraenen, A.; Van Lommel, L.; Lehnert, S.; et al. Tissue-specific disallowance of housekeeping genes: The other face of cell differentiation. Genome Res. 2011, 21, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar] [CrossRef]
- Ishihara, H.; Wang, H.Y.; Drewes, L.R.; Wollheim, C.B. Overexpression of monocarboxylate transporter and lactate dehydrogenase alters insulin secretory responses to pyruvate and lactate in beta cells. J. Clin. Investig. 1999, 104, 1621–1629. [Google Scholar] [CrossRef] [Green Version]
- Otonkoski, T.; Jiao, H.; Kaminen-Ahola, N.; Tapia-Paez, I.; Ullah, M.S.; Parton, L.E.; Schuit, F.; Quintens, R.; Sipilä, I.; Mayatepek, E.; et al. Physical Exercise–Induced Hypoglycemia Caused by Failed Silencing of Monocarboxylate Transporter 1 in Pancreatic beta Cells. Am. J. Hum. Genet. 2007, 81, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Sener, A.; Blachier, F.; Malaisse, W.J. Crabtree effect in tumoral pancreatic islet cells. J. Biol. Chem. 1988, 263, 1904–1909. [Google Scholar] [CrossRef]
- Hellman, B.; Idahl, L.A.; Sehlin, J.; Taljedal, I.B. Influence of anoxia on glucose metabolism in pancreatic islets: Lack of correlation between fructose-1,6-diphosphate and apparent glycolytic flux. Diabetologia 1975, 11, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaafar, R.; Tran, S.; Shah, A.N.; Sun, G.; Valdearcos, M.; Marchetti, P.; Masini, M.; Swisa, A.; Giacometti, S.; Bernal-Mizrachi, E.; et al. mTORC1-to-AMPK switching underlies beta cell metabolic plasticity during maturation and diabetes. J. Clin. Investig. 2019, 129, 4124–4137. [Google Scholar] [CrossRef] [Green Version]
- Pullen, T.J.; Xavier, G.D.S.; Kelsey, G.; Rutter, G.A. miR-29a and miR-29b Contribute to Pancreatic beta-Cell-Specific Silencing of Monocarboxylate Transporter 1 (Mct1). Mol. Cell. Biol. 2011, 31, 3182–3194. [Google Scholar] [CrossRef] [Green Version]
- Kaspi, H.; Pasvolsky, R.; Hornstein, E. Could microRNAs contribute to the maintenance of beta cell identity? Trends Endocrinol. Metab. 2014, 25, 285–292. [Google Scholar] [CrossRef]
- Sałówka, A.; Martinez-Sanchez, A. Molecular Mechanisms of Nutrient-Mediated Regulation of MicroRNAs in Pancreatic beta-cells. Front. Endocrinol. 2021, 12, 704824. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Sun, J.J.; Lin, Z.; Zhang, W.Z.; Wang, S.; Wang, W.Q.; Wang, Q.D.; Ning, G. m6A mRNA Methylation Controls Functional Maturation in Neonatal Murine beta-Cells. Diabetes 2020, 69, 1708–1722. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.A.; Bégin, J.; McDonald, C.L.; Vanderkruk, B.; Stephan, T.L.; Hoffman, B.G. H3K4 Trimethylation Is Required for Postnatal Pancreatic Endocrine Cell Functional Maturation. Diabetes 2021, 70, 2568–2579. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.Y.; Stein, G.H.; Pan, N.; Goebbels, S.; Hörnberg, H.; Nave, K.-A.; Herrera, P.; White, P.; Kaestner, K.H.; Sussel, L.; et al. Pancreatic beta Cells Require NeuroD to Achieve and Maintain Functional Maturity. Cell Metab. 2010, 11, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Blum, B.; Hrvatin, S.; Schuetz, C.; Bonal, C.; Rezania, A.; Melton, D.A. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin. Nat. Biotechnol. 2012, 30, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Helman, A.; Cangelosi, A.L.; Davis, J.C.; Pham, Q.; Rothman, A.; Faust, A.L.; Straubhaar, J.R.; Sabatini, D.M.; Melton, D.A. A Nutrient-Sensing Transition at Birth Triggers Glucose-Responsive Insulin Secretion. Cell Metab. 2020, 31, 1004–1016.e5. [Google Scholar] [CrossRef]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Sinagoga, K.L.; Stone, W.J.; Schiesser, J.V.; Schweitzer, J.I.; Sampson, L.; Zheng, Y.; Wells, J.M. Distinct roles for the mTOR pathway in postnatal morphogenesis, maturation and function of pancreatic islets. Development 2017, 144, 2402–2414. [Google Scholar] [CrossRef] [Green Version]
- Sakhneny, L.; Mueller, L.; Schonblum, A.; Azaria, S.; Burganova, G.; Epshtein, A.; Isaacson, A.; Wilson, H.; Spagnoli, F.M.; Landsman, L. The postnatal pancreatic microenvironment guides beta cell maturation through BMP4 production. Dev. Cell 2021, 56, 2703–2711.e5. [Google Scholar] [CrossRef]
- Gerst, F.; Kemter, E.; Lorza-Gil, E.; Kaiser, G.; Fritz, A.-K.; Nano, R.; Piemonti, L.; Gauder, M.; Dahl, A.; Nadalin, S.; et al. The hepatokine fetuin-A disrupts functional maturation of pancreatic beta cells. Diabetologia 2021, 64, 1358–1374. [Google Scholar] [CrossRef]
- Matsuda, H.; Mullapudi, S.T.; Zhang, Y.X.; Hesselson, D.; Stainier, D.Y.R. Thyroid Hormone Coordinates Pancreatic Islet Maturation During the Zebrafish Larval-to-Juvenile Transition to Maintain Glucose Homeostasis. Diabetes 2017, 66, 2623–2635. [Google Scholar] [CrossRef] [Green Version]
- Elsakr, J.M.; Dunn, J.C.; Tennant, K.; Zhao, S.K.; Kroeten, K.; Pasek, R.C.; Takahashi, D.L.; Dean, T.A.; Edwards, D.R.V.; McCurdy, C.E.; et al. Maternal Western-style diet affects offspring islet composition and function in a non-human primate model of maternal over-nutrition. Mol. Metab. 2019, 25, 73–82. [Google Scholar] [CrossRef]
- Yessoufou, A.; Moutairou, K. Maternal Diabetes in Pregnancy: Early and Long-Term Outcomes on the Offspring and the Concept of “Metabolic Memory”. Exp. Diabetes Res. 2011, 2011, 218598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christians, J.K.; Lennie, K.I.; Wild, L.K.; Garcha, R. Effects of high-fat diets on fetal growth in rodents: A systematic review. Reprod. Biol. Endocrinol. 2019, 17, 1–12. [Google Scholar] [CrossRef]
- Eberle, C.; Ament, C. A combined in vivo and in silico model shows specific predictors of individual trans-generational diabetic programming. J. Dev. Orig. Health Dis. 2021, 12, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Ryckman, K.K. Epigenetic and developmental influences on the risk of obesity, diabetes, and metabolic syndrome. Diabetes Metab. Syndr. Obes. Targets Ther. 2015, 8, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Bleker, L.S.; de Rooij, S.R.; Painter, R.C.; Ravelli, A.C.J.; Roseboom, T.J. Cohort profile: The Dutch famine birth cohort (DFBC)—A prospective birth cohort study in the Netherlands. BMJ Open 2021, 11, e042078. [Google Scholar] [CrossRef]
- de Rooij, S.R.; Roseboom, T.J.; Painter, R.C. Famines in the Last 100 Years: Implications for Diabetes. Curr. Diabetes Rep. 2014, 14, 1–10. [Google Scholar] [CrossRef]
- Forsén, T.; Eriksson, J.; Tuomilehto, J.; Reunanen, A.; Osmond, C.; Barker, D. The Fetal and Childhood Growth of Persons Who Develop Type 2 Diabetes. Ann. Intern. Med. 2000, 133, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Thorn, S.R.; Rozance, P.J.; Brown, L.D.; Hay, W.W., Jr. The Intrauterine Growth Restriction Phenotype: Fetal Adaptations and Potential Implications for Later Life Insulin Resistance and Diabetes. Semin. Reprod. Med. 2011, 29, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Chelbi, S.T.; Doridot, L.; Mondon, F.; Dussour, C.; Rebourcet, R.; Busato, F.; Gascoin-Lachambre, G.; Barbaux, S.; Rigourd, V.; Mignot, T.-M.; et al. Combination of promoter hypomethylation and PDX1 overexpression leads toTBX15decrease in vascular IUGR placentas. Epigenetics 2011, 6, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Economides, D.L.; Nicolaides, K.H.; Campbell, S. Metabolic and endocrine findings in appropriate and small for gestational age fetuses. J. Périnat. Med. 1991, 19, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J.P.; Clark, P.M.S.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, U.; Hubinont, C.; Santolaya, J.; Fisk, N.M.; Rodeck, C.H. Effects of Fetal Intravenous Glucose Challenge in Normal and Growth Retarded Fetuses. Horm. Metab. Res. 1990, 22, 426–430. [Google Scholar] [CrossRef]
- Nobili, V.; Marcellini, M.; Marchesini, G.; Vanni, E.; Manco, M.; Villani, A.; Bugianesi, E. Intrauterine Growth Retardation, Insulin Resistance, and Nonalcoholic Fatty Liver Disease in Children. Diabetes Care 2007, 30, 2638–2640. [Google Scholar] [CrossRef] [Green Version]
- Van Assche, F.A.; De Prins, F.; Aerts, L.; Verjans, M. Endocrine pancreas in small-for-dates infants. BJOG Int. J. Obstet. Gynaecol. 1977, 84, 751–753. [Google Scholar] [CrossRef]
- Barisione, M.; Carlini, F.; Gradaschi, R.; Camerini, G.; Adami, G.F. Body weight at developmental age in siblings born to mothers before and after surgically induced weight loss. Surg. Obes. Relat. Dis. 2012, 8, 387–391. [Google Scholar] [CrossRef]
- Boney, C.M.; Verma, A.; Tucker, R.; Vohr, B.R. Metabolic Syndrome in Childhood: Association with Birth Weight, Maternal Obesity, and Gestational Diabetes Mellitus. Pediatrics 2005, 115, e290–e296. [Google Scholar] [CrossRef] [Green Version]
- Catalano, P.M.; Presley, L.; Minium, J.; de Mouzon, S.H. Fetuses of Obese Mothers Develop Insulin Resistance in Utero. Reprod. Sci. 2009, 32, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Hochner, H.; Friedlander, Y.; Calderon-Margalit, R.; Meiner, V.; Sagy, Y.; Avgil-Tsadok, M.; Burger, A.; Savitsky, B.; Siscovick, D.S.; Manor, O. Associations of Maternal Prepregnancy Body Mass Index and Gestational Weight Gain with Adult Offspring Cardiometabolic Risk Factors the Jerusalem Perinatal Family Follow-Up Study. Circulation 2012, 125, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, S.M.; Adams, A.K.; Prince, R.J. Early predictors of obesity and cardiovascular risk among American Indian children. Matern. Child Health J. 2012, 16, 1879–1886. [Google Scholar] [CrossRef] [Green Version]
- Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; McIntyre, H.D.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [PubMed] [Green Version]
- Mingrone, G.; Manco, M.; Mora, M.E.V.; Guidone, C.; Iaconelli, A.; Gniuli, D.; Leccesi, L.; Chiellini, C.; Ghirlanda, G. Influence of Maternal Obesity on Insulin Sensitivity and Secretion in Offspring. Diabetes Care 2008, 31, 1872–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, R.M.; Allan, K.M.; Raja, E.A.; Bhattacharya, S.; McNeill, G.; Hannaford, P.C.; Sarwar, N.; Lee, A.J.; Bhattacharya, S.; Norman, J.E. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: Follow-up of 1,323,275 person years. BMJ 2013, 347, f4539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemond, J.; Robbins, R.B.; Young, P.C. The Effects of Maternal Obesity on Neonates, Infants, Children, Adolescents, and Adults. Clin. Obstet. Gynecol. 2016, 59, 216–227. [Google Scholar] [CrossRef]
- Sweeting, A.; Wong, J.; Murphy, H.R.; Ross, G.P. A clinical update on Gestational Diabetes Mellitus. Endocr. Rev. 2022, bnac003. [Google Scholar] [CrossRef]
- Elsakr, J.M.; Pasek, R.C.; Takahashi, D.; Grove, K.L.; Powers, A.C.; Gannon, M.A. The Effects of In Utero High-Fat Diet Exposure on the Endocrine Pancreas of the Offspring. Diabetes 2017, 66, A574–A575. [Google Scholar]
- Elsakr, J.M.; Gannon, M. Developmental programming of the pancreatic islet by in utero overnutrition. Trends Dev. Biol. 2017, 10, 79–95. [Google Scholar]
- Nielsen, J.H.; Haase, T.N.; Jaksch, C.; Nalla, A.; Søstrup, B.; Nalla, A.A.; Larsen, L.; Rasmussen, M.; Dalgaard, L.T.; Gaarn, L.W.; et al. Impact of fetal and neonatal environment on beta cell function and development of diabetes. Acta Obstet. Gynecol. Scand. 2014, 93, 1109–1122. [Google Scholar] [CrossRef]
- Drigo, R.A.E.; Ali, Y.; Diez, J.; Srinivasan, D.K.; Berggren, P.-O.; Boehm, B.O. New insights into the architecture of the islet of Langerhans: A focused cross-species assessment. Diabetologia 2015, 58, 2218–2228. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, L.M.; Nagao, M.; Kusinski, L.C.; Fernandez-Twinn, D.S.; Eliasson, L.; Ozanne, S.E. Exposure to maternal obesity programs sex differences in pancreatic islets of the offspring in mice. Diabetologia 2020, 63, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Graus-Nunes, F.; Frantz, E.D.; Lannes, W.R.; Menezes, M.C.D.S.; Mandarim-De-Lacerda, C.A.; Souza-Mello, V. Pregestational maternal obesity impairs endocrine pancreas in male F1 and F2 progeny. Nutrition 2015, 31, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, L.; Wang, Z.W.; Zhang, J.Q. Maternal high-fat diet regulates glucose metabolism and pancreatic beta cell phenotype in mouse offspring at weaning. PeerJ 2020, 8, e9407. [Google Scholar] [CrossRef] [PubMed]
- Tuohetimulati, G.; Uchida, T.; Toyofuku, Y.; Abe, H.; Fujitani, Y.; Hirose, T.; Takeda, S.; Watada, H. Effect of maternal high-fat diet on pancreatic beta cells of the offspring. Diabetol. Int. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Ye, T.-T.; Liu, C.-X.; Wang, L.; Chen, Y.-W.; Dong, Y. Maternal high-fat diet impairs glucose metabolism, beta-cell function and proliferation in the second generation of offspring rats. Nutr. Metab. 2017, 14, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Xu, J.; Epstein, F.N.; Liu, Y.Q. Long-term effect of maternal obesity on pancreatic beta cells of offspring: Reduced beta cell adaptation to high glucose and high-fat diet challenges in adult female mouse offspring. Diabetologia 2005, 48, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Brar, N.; Morriseau, T.S.; Kereliuk, S.M.; Fonseca, M.A.; Cole, L.K.; Jha, A.; Xiang, B.; Hunt, K.L.; Seshadri, N.; et al. Gestational Diabetes Adversely Affects Pancreatic Islet Architecture and Function in the Male Rat Offspring. Endocrinology 2019, 160, 1907–1925. [Google Scholar] [CrossRef]
- Ng, S.-F.; Lin, R.C.Y.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef]
- Russell, M.A.; Morgan, N.G. The impact of anti-inflammatory cytokines on the pancreatic beta-cell. Islets 2014, 6, e950547. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Shyh-Chang, N.; Segre, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 Axis Regulates Glucose Metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, T.D.; Ingerslev, L.R.; Alm, P.S.; Versteyhe, S.; Massart, J.; Rasmussen, M.; Donkin, I.; Sjogren, R.; Mudry, J.M.; Vetterli, L.; et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2016, 5, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Megel, C.; Morelle, G.; Lalande, S.; Duchêne, A.-M.; Small, I.; Maréchal-Drouard, L. Surveillance and Cleavage of Eukaryotic tRNAs. Int. J. Mol. Sci. 2015, 16, 1873–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Yan, M.H.; Cao, Z.H.; Li, X.; Zhang, Y.F.; Shi, J.C.; Feng, G.-H.; Peng, H.Y.; Zhang, X.D.; Zhang, Y.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Chen, C.-H.; Hu, C.; Long, J.R.; Ong, R.T.H.; Sim, X.L.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat. Genet. 2011, 44, 67–72. [Google Scholar] [CrossRef]
- Blue, E.K.; Ballman, K.; Boyle, F.; Oh, E.; Kono, T.; Quinney, S.K.; Thurmond, D.C.; Evans-Molina, C.; Haneline, L.S. Fetal hyperglycemia and a high-fat diet contribute to aberrant glucose tolerance and hematopoiesis in adult rats. Pediatr. Res. 2015, 77, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, M.; Katewa, S.D.; Palaniyappan, A.; Pandya, J.D.; Patel, M.S. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol.-Endoc. Metab. 2006, 291, E792–E799. [Google Scholar] [CrossRef]
- Ortega, Á.; Berná, G.; Rojas, A.; Martín, F.; Soria, B. Gene-Diet Interactions in Type 2 Diabetes: The Chicken and Egg Debate. Int. J. Mol. Sci. 2017, 18, 1188. [Google Scholar] [CrossRef]
- Kollée, L.A.A.; Monnens, L.A.H.; Trijbels, J.M.F.; Veerkamp, J.H.; Janssen, A.J.M. Experimental intrauterine growth retardation in the rat. Evaluation of the Wigglesworth model. Early Hum. Dev. 1979, 3, 295–300. [Google Scholar] [CrossRef]
- Su, Y.T.; Jiang, X.L.; Li, Y.L.; Li, F.; Cheng, Y.L.; Peng, Y.; Song, D.L.; Hong, J.; Ning, G.; Cao, Y.; et al. Maternal Low Protein Isocaloric Diet Suppresses Pancreatic beta-Cell Proliferation in Mouse Offspring via miR-15b. Endocrinology 2016, 157, 4782–4793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theys, N.; Clippe, A.; Bouckenooghe, T.; Reusens, B.; Remacle, C. Early Low Protein Diet Aggravates Unbalance between Antioxidant Enzymes Leading to Islet Dysfunction. PLoS ONE 2009, 4, e6110. [Google Scholar] [CrossRef] [PubMed]
- Haase, T.N.; Rasmussen, M.; Jaksch, C.A.M.; Gaarn, L.W.; Petersen, C.K.; Billestrup, N.; Nielsen, J.H. Growth arrest specific protein (GAS) 6: A role in the regulation of proliferation and functional capacity of the perinatal rat beta cell. Diabetologia 2013, 56, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Petrik, J.; Reusens, B.; Arany, E.; Remacle, C.; Coelho, C.; Hoet, J.J.; Hill, D.J. A Low Protein Diet Alters the Balance of Islet Cell Replication and Apoptosis in the Fetal and Neonatal Rat and Is Associated with a Reduced Pancreatic Expression of Insulin-Like Growth Factor-II. Endocrinology 1999, 140, 4861–4873. [Google Scholar] [CrossRef] [PubMed]
- Rashid, C.S.; Lien, Y.-C.; Bansal, A.; Jaeckle-Santos, L.J.; Li, C.; Won, K.J.; Simmons, R.A. Transcriptomic Analysis Reveals Novel Mechanisms Mediating Islet Dysfunction in the Intrauterine Growth–Restricted Rat. Endocrinology 2018, 159, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumortier, O.; Hinault, C.; Gautier, N.; Patouraux, S.; Casamento, V.; Van Obberghen, E. Maternal Protein Restriction Leads to Pancreatic Failure in Offspring: Role of Misexpressed MicroRNA-375. Diabetes 2014, 63, 3416–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandro, E.u.; Gregg, B.; Wallen, T.; Kumusoglu, D.; Meister, D.; Chen, A.; Merrins, M.J.; Satin, L.S.; Liu, M.; Arvan, P.; et al. Maternal diet–induced microRNAs and mTOR underlie beta cell dysfunction in offspring. J. Clin. Investig. 2014, 124, 4395–4410. [Google Scholar] [CrossRef] [Green Version]
- El Ouaamari, A.; Baroukh, N.; Martens, G.A.; Lebrun, P.; Pipeleers, D.; van Obberghen, E. miR-375 targets 3′-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes 2008, 57, 2708–2717. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Trejo, A.; Ortiz-López, M.G.; Zambrano, E.; Granados-Silvestre, M.D.; Méndez, C.; Blondeau, B.; Bréant, B.; Nathanielsz, P.W.; Menjívar, M. Developmental programming of neonatal pancreatic beta-cells by a maternal low-protein diet in rats involves a switch from proliferation to differentiation. Am. J. Physiol.-Endoc. Metab. 2012, 302, E1431–E1439. [Google Scholar] [CrossRef] [Green Version]
- Reusens, B.; Theys, N.; Dumortier, O.; Goosse, K.; Remacle, C. Maternal malnutrition programs the endocrine pancreas in progeny. Am. J. Clin. Nutr. 2011, 94, 1824S–1829S. [Google Scholar] [CrossRef]
- Sparre, T.; Reusens, B.; Cherif, H.; Larsen, M.R.; Roepstorff, P.; Fey, S.J.; Larsen, P.M.; Remacle, C.; Nerup, J. Intrauterine programming of fetal islet gene expression in rats-effects of maternal protein restriction during gestation revealed by proteome analysis. Diabetologia 2003, 46, 1497–1511. [Google Scholar] [CrossRef] [Green Version]
- Reusens, B.; Sparre, T.; Kalbe, L.; Bouckenooghe, T.; Theys, N.; Kruhøffer, M.; Ørntoft, T.F.; Nerup, J.; Remacle, C. The intrauterine metabolic environment modulates the gene expression pattern in fetal rat islets: Prevention by maternal taurine supplementation. Diabetologia 2008, 51, 836–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, J.Y.; Liu, C.Y.; Naji, A.; Stoffers, D.A. MicroRNA-7 Regulates the mTOR Pathway and Proliferation in Adult Pancreatic beta-Cells. Diabetes 2013, 62, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, J.C.; Miranda, R.A.; Barella, L.F.; Torrezan, R.; Agostinho, A.R.; Ribeiro, T.A.S.; Franco, C.C.S.; Malta, A.; Tófolo, L.P.; Gravena, C.; et al. Impaired beta-cell function in the adult offspring of rats fed a protein-restricted diet during lactation is associated with changes in muscarinic acetylcholine receptor subtypes. Br. J. Nutr. 2014, 111, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.F.; Fazzari, M.J.; Niu, H.S.; Barzilai, N.; Simmons, R.A.; Greally, J.M. Experimental Intrauterine Growth Restriction Induces Alterations in DNA Methylation and Gene Expression in Pancreatic Islets of Rats. J. Biol. Chem. 2010, 285, 15111–15118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx. J. Clin. Investig. 2008, 118, 2316–2324. [Google Scholar] [CrossRef] [Green Version]
- Sandovici, I.; Smith, N.H.; Nitert, M.D.; Ackers-Johnson, M.; Uribe-Lewis, S.; Ito, Y.; Jones, R.H.; Marquez, V.E.; Cairns, W.; Tadayyon, M.; et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc. Natl. Acad. Sci. USA 2011, 108, 5449–5454. [Google Scholar] [CrossRef] [Green Version]
- Silander, K.; Mohlke, K.L.; Scott, L.J.; Peck, E.C.; Hollstein, P.; Skol, A.D.; Jackson, A.U.; Deloukas, P.; Hunt, S.; Stavrides, G.; et al. Genetic Variation Near the Hepatocyte Nuclear Factor-4alpha Gene Predicts Susceptibility to Type 2 Diabetes. Diabetes 2004, 53, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Vatamaniuk, M.Z.; Lee, C.S.; Flaschen, R.C.; Fulmer, J.T.; Matschinsky, F.M.; Duncan, S.A.; Kaestner, K.H. The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J. Clin. Investig. 2005, 115, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Chamson-Reig, A.; Arany, E.J.; Summers, K.; Hill, D.J. A low protein diet in early life delays the onset of diabetes in the non-obese diabetic mouse. J. Endocrinol. 2009, 201, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Theys, N.; Bouckenooghe, T.; Ahn, M.-T.; Remacle, C.; Reusens, B. Maternal low-protein diet alters pancreatic islet mitochondrial function in a sex-specific manner in the adult rat. Am. J. Physiol. Integr. Comp. Physiol. 2009, 297, R1516–R1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Maternal Influence | Clinical Signs of the Progeny at Birth and during Childhood | Clinical Signs of the Progeny during Adulthood | References |
|---|---|---|---|
| Famine during pregnancy | ➢ Thinness and low weight at birth | ➢ Increased body mass index and increased risk for obesity ➢ Reduced lean body mass ➢ Metabolic syndrome ➢ Increased risk for insulin resistance ➢ Glucose intolerance ➢ High prevalence of T2D ➢ Early onset of coronary heart disease | [57,58,59,60] |
| Pathological pregnancy with intrauterine growth restriction * | ➢ Placental epigenetic marks of PDX1 target genes involved in glucose-dependent regulation of the insulin gene expression; correlation of altered epigenetic events with reduced body weight of the newborns ➢ Reduced lean mass and increased percentage body fat up to 2 years of age ➢ Insulin resistance ➢ Reduced transplacental transport of amino acids ➢ Impaired pancreatic islet function and systemic insulin release ➢ Reduced fetal endocrine pancreatic tissue and insulin producing β-cells ➢ Hypoinsulinemia | ➢ Persistently reduced lean body mass ➢ Glucose intolerance | [60,61,62,63,64,65,66] |
| Maternal obesity | ➢ Increased body weight at birth and at puberty ➢ Increased incidence of overweight and obesity ➢ Increased insulin resistance at up to 20 years of age ➢ Fetal hyperinsulinemia | ➢ Increased body weight ➢ Increased incidence of overweight and obesity ➢ Lower circulating high density lipoproteins ➢ 29% increased risk of death caused by cardiovascular disease | [67,68,69,70,71,72,73,74] |
| Gestation diabetes mellitus | ➢ Macrosomia ➢ Hypoglycemia ➢ Infancy obesity | ➢ Metabolic syndrome ➢ Excess abdominal adiposity ➢ Hyperinsulinemia ➢ Dysglycemia ➢ Obesity ➢ T2D ➢ Cardiova6scular disease | [75,76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacovetti, C.; Regazzi, R. Mechanisms Underlying the Expansion and Functional Maturation of β-Cells in Newborns: Impact of the Nutritional Environment. Int. J. Mol. Sci. 2022, 23, 2096. https://doi.org/10.3390/ijms23042096
Jacovetti C, Regazzi R. Mechanisms Underlying the Expansion and Functional Maturation of β-Cells in Newborns: Impact of the Nutritional Environment. International Journal of Molecular Sciences. 2022; 23(4):2096. https://doi.org/10.3390/ijms23042096
Chicago/Turabian StyleJacovetti, Cécile, and Romano Regazzi. 2022. "Mechanisms Underlying the Expansion and Functional Maturation of β-Cells in Newborns: Impact of the Nutritional Environment" International Journal of Molecular Sciences 23, no. 4: 2096. https://doi.org/10.3390/ijms23042096