
Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs

 , , , , , ,
, , , , , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
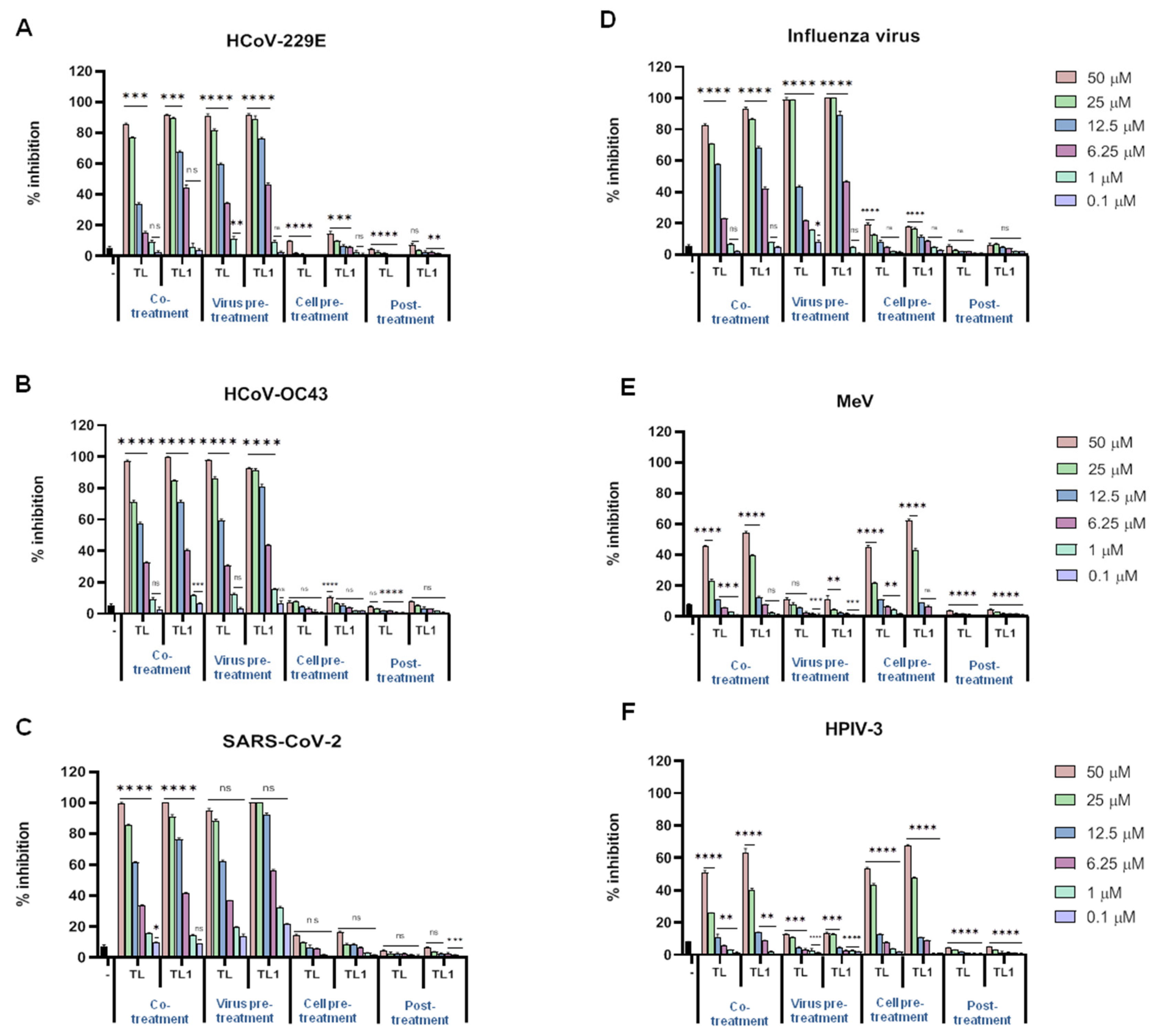
2.1. Native Temporin L and Its Analog ([Pro3, DLeu9]TL) Antiviral Activities
2.2. Cytotoxicity of Native Temporin L and Its Analog ([Pro3, DLeu9]TL)
2.3. Cytotoxicity and Antiviral Activities of Gly10-Replaced TL1 Analogues
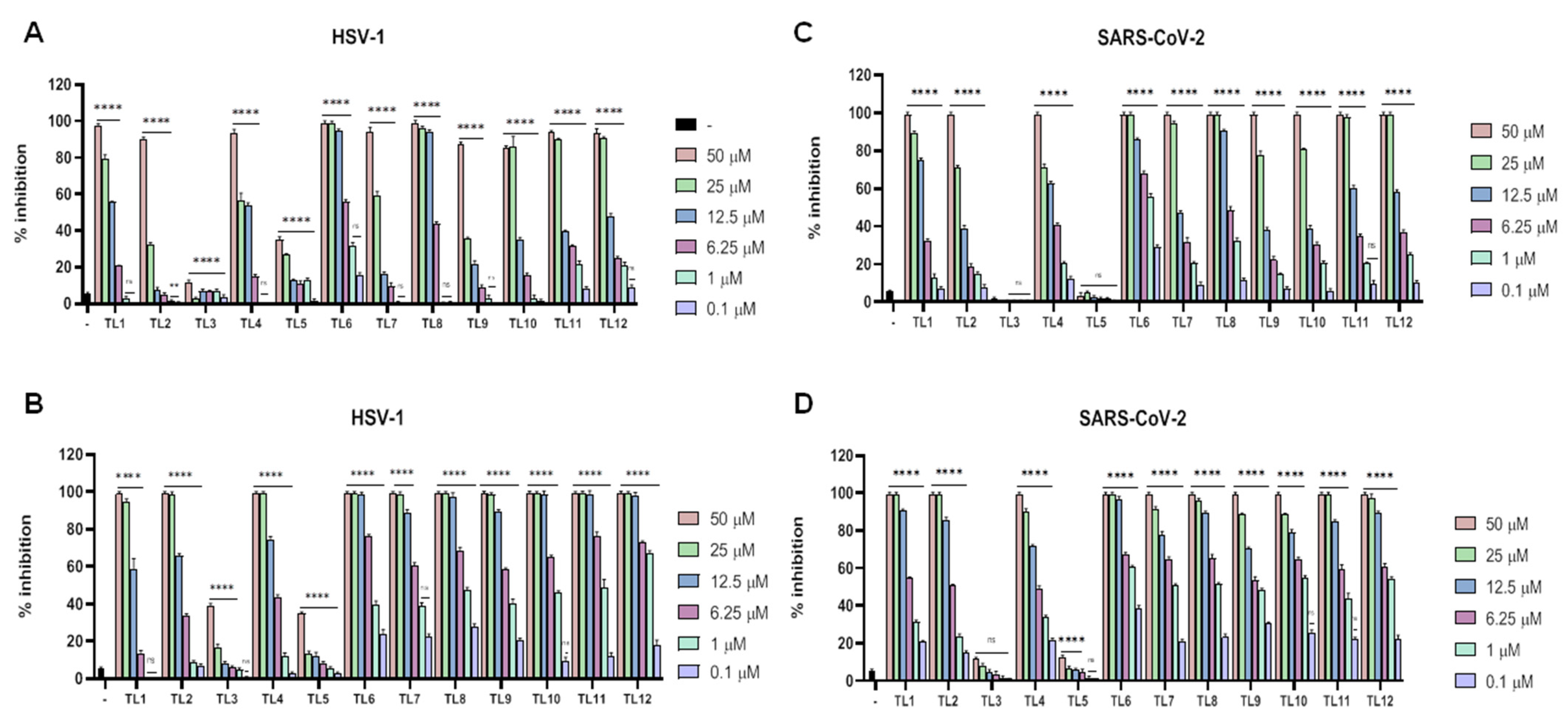
2.4. Cytotoxicity and Antiviral Activities of TL6 and Its Lipid-Conjugates
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Materials
4.1.2. Peptide Synthesis
4.1.3. Conjugation of Cholesterol to TL6.1–TL6.4
4.1.4. Conjugation of Fatty Acids to TL6.5–TL6.8
4.2. Biology
4.2.1. Cell and Virus Culture
4.2.2. Cytotoxicity
4.2.3. Hemolytic Assays
4.2.4. Antiviral Activity
4.2.5. Calculation of Therapeutic Index
4.2.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Clercq, E. Fifty Years in Search of Selective Antiviral Drugs. J. Med. Chem. 2019, 62, 7322–7339. [Google Scholar] [CrossRef] [PubMed]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, S.; Symons, J.A.; Deval, J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antivir. Res. 2018, 155, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Devnath, P.; Masud, H. Nipah virus: A potential pandemic agent in the context of the current severe acute respiratory syndrome coronavirus 2 pandemic. New Microbes New Infect. 2021, 41, 100873. [Google Scholar] [CrossRef] [PubMed]
- Huchting, J. Targeting viral genome synthesis as broad-spectrum approach against RNA virus infections. Antivir. Chem. Chemother. 2020, 28, 2040206620976786. [Google Scholar] [CrossRef] [PubMed]
- Artese, A.; Svicher, V.; Costa, G.; Salpini, R.; Di Maio, V.C.; Alkhatib, M.; Ambrosio, F.A.; Santoro, M.M.; Assaraf, Y.G.; Alcaro, S.; et al. Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses. Drug Resist. Updat. 2020, 53, 100721. [Google Scholar] [CrossRef]
- Pattnaik, G.P.; Chakraborty, H. Entry Inhibitors: Efficient Means to Block Viral Infection. J. Membr. Biol. 2020, 253, 425–444. [Google Scholar] [CrossRef]
- Hsieh, I.N.; Hartshorn, K.L. The Role of Antimicrobial Peptides in Influenza Virus Infection and Their Potential as Antiviral and Immunomodulatory Therapy. Pharmaceuticals 2016, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Cheshenko, N.; Lehrer, R.I.; Herold, B.C. NP-1, a rabbit alpha-defensin, prevents the entry and intercellular spread of herpes simplex virus type 2. Antimicrob. Agents Chemother. 2003, 47, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Carriel-Gomes, M.C.; Kratz, J.M.; Barracco, M.A.; Bachere, E.; Barardi, C.R.; Simoes, C.M. In vitro antiviral activity of antimicrobial peptides against herpes simplex virus 1, adenovirus, and rotavirus. Mem. Inst. Oswaldo Cruz 2007, 102, 469–472. [Google Scholar] [CrossRef]
- Yasin, B.; Pang, M.; Turner, J.S.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I.; Wagar, E.A. Evaluation of the inactivation of infectious Herpes simplex virus by host-defense peptides. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Gwyer Findlay, E.; Currie, S.M.; Davidson, D.J. Cationic host defence peptides: Potential as antiviral therapeutics. BioDrugs 2013, 27, 479–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galdiero, S.; Falanga, A.; Tarallo, R.; Russo, L.; Galdiero, E.; Cantisani, M.; Morelli, G.; Galdiero, M. Peptide inhibitors against herpes simplex virus infections. J. Pept. Sci. 2013, 19, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Vilas Boas, L.C.P.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Varga, J.F.A.; Bui-Marinos, M.P.; Katzenback, B.A. Frog Skin Innate Immune Defences: Sensing and Surviving Pathogens. Front. Immunol. 2018, 9, 3128. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Shai, Y. Temporins and their synergism against Gram-negative bacteria and in lipopolysaccharide detoxification. Biochim. Biophys. Acta 2009, 1788, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Di Grazia, A.; Cappiello, F.; Imanishi, A.; Mastrofrancesco, A.; Picardo, M.; Paus, R.; Mangoni, M.L. The Frog Skin-Derived Antimicrobial Peptide Esculentin-1a(1-21)NH2 Promotes the Migration of Human HaCaT Keratinocytes in an EGF Receptor-Dependent Manner: A Novel Promoter of Human Skin Wound Healing? PLoS ONE 2015, 10, e0128663. [Google Scholar] [CrossRef] [Green Version]
- Conlon, J.M. Structural diversity and species distribution of host-defense peptides in frog skin secretions. Cell Mol. Life Sci. 2011, 68, 2303–2315. [Google Scholar] [CrossRef]
- Rinaldi, A.C. Antimicrobial peptides from amphibian skin: An expanding scenario. Curr. Opin. Chem. Biol. 2002, 6, 799–804. [Google Scholar] [CrossRef]
- Ladram, A.; Nicolas, P. Antimicrobial peptides from frog skin: Biodiversity and therapeutic promises. Front. Biosci. Landmark Ed. 2016, 21, 1341–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egal, M.; Conrad, M.; MacDonald, D.L.; Maloy, W.L.; Motley, M.; Genco, C.A. Antiviral effects of synthetic membrane-active peptides on herpes simplex virus, type 1. Int. J. Antimicrob. Agents 1999, 13, 57–60. [Google Scholar] [CrossRef]
- Dean, R.E.; O’Brien, L.M.; Thwaite, J.E.; Fox, M.A.; Atkins, H.; Ulaeto, D.O. A carpet-based mechanism for direct antimicrobial peptide activity against vaccinia virus membranes. Peptides 2010, 31, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Belaid, A.; Aouni, M.; Khelifa, R.; Trabelsi, A.; Jemmali, M.; Hani, K. In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J. Med. Virol. 2002, 66, 229–234. [Google Scholar] [CrossRef]
- Bergaoui, I.; Zairi, A.; Tangy, F.; Aouni, M.; Selmi, B.; Hani, K. In vitro antiviral activity of dermaseptin S(4) and derivatives from amphibian skin against herpes simplex virus type 2. J. Med. Virol. 2013, 85, 272–281. [Google Scholar] [CrossRef]
- Lorin, C.; Saidi, H.; Belaid, A.; Zairi, A.; Baleux, F.; Hocini, H.; Belec, L.; Hani, K.; Tangy, F. The antimicrobial peptide dermaseptin S4 inhibits HIV-1 infectivity in vitro. Virology 2005, 334, 264–275. [Google Scholar] [CrossRef]
- Mechlia, M.B.; Belaid, A.; Castel, G.; Jallet, C.; Mansfield, K.L.; Fooks, A.R.; Hani, K.; Tordo, N. Dermaseptins as potential antirabies compounds. Vaccine 2019, 37, 4694–4700. [Google Scholar] [CrossRef]
- Monteiro, J.M.C.; Oliveira, M.D.; Dias, R.S.; Nacif-Marcal, L.; Feio, R.N.; Ferreira, S.O.; Oliveira, L.L.; Silva, C.C.; Paula, S.O. The antimicrobial peptide HS-1 inhibits dengue virus infection. Virology 2018, 514, 79–87. [Google Scholar] [CrossRef]
- Holthausen, D.J.; Lee, S.H.; Kumar, V.T.; Bouvier, N.M.; Krammer, F.; Ellebedy, A.H.; Wrammert, J.; Lowen, A.C.; George, S.; Pillai, M.R.; et al. An Amphibian Host Defense Peptide Is Virucidal for Human H1 Hemagglutinin-Bearing Influenza Viruses. Immunity 2017, 46, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Simmaco, M.; Mignogna, G.; Canofeni, S.; Miele, R.; Mangoni, M.L.; Barra, D. Temporins, antimicrobial peptides from the European red frog Rana temporaria. Eur. J. Biochem. 1996, 242, 788–792. [Google Scholar] [CrossRef]
- Mangoni, M.L. Temporins, anti-infective peptides with expanding properties. Cell Mol. Life Sci. 2006, 63, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Grazia, A.D.; Cappiello, F.; Casciaro, B.; Luca, V. Naturally Occurring Peptides from Rana temporaria: Antimicrobial Properties and More. Curr. Top. Med. Chem. 2016, 16, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Marcocci, M.E.; Amatore, D.; Villa, S.; Casciaro, B.; Aimola, P.; Franci, G.; Grieco, P.; Galdiero, M.; Palamara, A.T.; Mangoni, M.L.; et al. The Amphibian Antimicrobial Peptide Temporin B Inhibits In Vitro Herpes Simplex Virus 1 Infection. Antimicrob. Agents Chemother. 2018, 62, e02367-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinchar, V.G.; Bryan, L.; Silphadaung, U.; Noga, E.; Wade, D.; Rollins-Smith, L. Inactivation of viruses infecting ectothermic animals by amphibian and piscine antimicrobial peptides. Virology 2004, 323, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, Z.; Andre, S.; Abbassi, F.; Humblot, V.; Lequin, O.; Bouceba, T.; Correia, I.; Casale, S.; Foulon, T.; Sereno, D.; et al. Insight into the mechanism of action of temporin-SHa, a new broad-spectrum antiparasitic and antibacterial agent. PLoS ONE 2017, 12, e0174024. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.; Lebeau, L.; Chessa, C.; Damour, A.; Ladram, A.; Oury, B.; Boutolleau, D.; Bodet, C.; Leveque, N. Comparison of Anti-Viral Activity of Frog Skin Anti-Microbial Peptides Temporin-Sha and [K(3)]SHa to LL-37 and Temporin-Tb against Herpes Simplex Virus Type 1. Viruses 2019, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, M.; Casciaro, B.; Genovese, A.; Brancaccio, D.; Marcocci, M.E.; Novellino, E.; Carotenuto, A.; Palamara, A.T.; Mangoni, M.L.; Nencioni, L. Temporin G, an amphibian antimicrobial peptide against influenza and parainfluenza respiratory viruses: Insights into biological activity and mechanism of action. FASEB J. 2021, 35, e21358. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Rinaldi, A.C.; Di Giulio, A.; Mignogna, G.; Bozzi, A.; Barra, D.; Simmaco, M. Structure-function relationships of temporins, small antimicrobial peptides from amphibian skin. Eur. J. Biochem. 2000, 267, 1447–1454. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, A.C.; Mangoni, M.L.; Rufo, A.; Luzi, C.; Barra, D.; Zhao, H.; Kinnunen, P.K.; Bozzi, A.; Di Giulio, A.; Simmaco, M. Temporin L: Antimicrobial, haemolytic and cytotoxic activities, and effects on membrane permeabilization in lipid vesicles. Biochem. J. 2002, 368, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Carotenuto, A.; Malfi, S.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.; Mangoni, M.L.; Gaddi, L.M.; Novellino, E.; Grieco, P. A different molecular mechanism underlying antimicrobial and hemolytic actions of temporins A and L. J. Med. Chem. 2008, 51, 2354–2362. [Google Scholar] [CrossRef]
- Saviello, M.R.; Malfi, S.; Campiglia, P.; Cavalli, A.; Grieco, P.; Novellino, E.; Carotenuto, A. New insight into the mechanism of action of the temporin antimicrobial peptides. Biochemistry 2010, 49, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Groot, H.; Munoz-Camargo, C.; Moscoso, J.; Riveros, G.; Salazar, V.; Kaston Florez, F.; Mitrani, E. Skin micro-organs from several frog species secrete a repertoire of powerful antimicrobials in culture. J. Antibiot. 2012, 65, 461–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zannella, C.; Mosca, F.; Mariani, F.; Franci, G.; Folliero, V.; Galdiero, M.; Tiscar, P.G.; Galdiero, M. Microbial Diseases of Bivalve Mollusks: Infections, Immunology and Antimicrobial Defense. Mar Drugs 2017, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.; Silberring, J.; Soliymani, R.; Heikkinen, S.; Kilpelainen, I.; Lankinen, H.; Kuusela, P. Antibacterial activities of temporin A analogs. FEBS Lett 2000, 479, 6–9. [Google Scholar] [CrossRef]
- Popovic, S.; Urban, E.; Lukic, M.; Conlon, J.M. Peptides with antimicrobial and anti-inflammatory activities that have therapeutic potential for treatment of acne vulgaris. Peptides 2012, 34, 275–282. [Google Scholar] [CrossRef]
- Grieco, P.; Carotenuto, A.; Auriemma, L.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.M.; Marcellini, L.; Luca, V.; Barra, D.; Novellino, E.; et al. The effect of d-amino acid substitution on the selectivity of temporin L towards target cells: Identification of a potent anti-Candida peptide. Biochim. Biophys. Acta 2013, 1828, 652–660. [Google Scholar] [CrossRef]
- Buommino, E.; Carotenuto, A.; Antignano, I.; Bellavita, R.; Casciaro, B.; Loffredo, M.R.; Merlino, F.; Novellino, E.; Mangoni, M.L.; Nocera, F.P.; et al. The Outcomes of Decorated Prolines in the Discovery of Antimicrobial Peptides from Temporin-L. ChemMedChem 2019, 14, 1283–1290. [Google Scholar] [CrossRef]
- Merlino, F.; Carotenuto, A.; Casciaro, B.; Martora, F.; Loffredo, M.R.; Di Grazia, A.; Yousif, A.M.; Brancaccio, D.; Palomba, L.; Novellino, E.; et al. Glycine-replaced derivatives of [Pro(3),DLeu(9)]TL, a temporin L analogue: Evaluation of antimicrobial, cytotoxic and hemolytic activities. Eur. J. Med. Chem. 2017, 139, 750–761. [Google Scholar] [CrossRef]
- Outlaw, V.K.; Bovier, F.T.; Mears, M.C.; Cajimat, M.N.; Zhu, Y.; Lin, M.J.; Addetia, A.; Lieberman, N.A.P.; Peddu, V.; Xie, X.; et al. Inhibition of Coronavirus Entry In Vitro and Ex Vivo by a Lipid-Conjugated Peptide Derived from the SARS-CoV-2 Spike Glycoprotein HRC Domain. mBio 2020, 11, e01935-20. [Google Scholar] [CrossRef]
- de Vries, R.D.; Schmitz, K.S.; Bovier, F.T.; Predella, C.; Khao, J.; Noack, D.; Haagmans, B.L.; Herfst, S.; Stearns, K.N.; Drew-Bear, J.; et al. Intranasal fusion inhibitory lipopeptide prevents direct-contact SARS-CoV-2 transmission in ferrets. Science 2021, 371, 1379–1382. [Google Scholar] [CrossRef]
- Luteijn, R.D.; Praest, P.; Thiele, F.; Sadasivam, S.M.; Singethan, K.; Drijfhout, J.W.; Bach, C.; de Boer, S.M.; Lebbink, R.J.; Tao, S.; et al. A Broad-Spectrum Antiviral Peptide Blocks Infection of Viruses by Binding to Phosphatidylserine in the Viral Envelope. Cells 2020, 9, 1989. [Google Scholar] [CrossRef] [PubMed]
- Brice, D.C.; Diamond, G. Antiviral Activities of Human Host Defense Peptides. Curr. Med. Chem. 2020, 27, 1420–1443. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.I.; Pham, T.K.; Kim, D.; Park, M.; Kim, B.O.; Cho, Y.H.; Kim, Y.W.; Lee, C. Identification of brevinin-1EMa-derived stapled peptides as broad-spectrum virus entry blockers. Virology 2021, 561, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhou, J.; Zhang, K.; Chu, H.; Liu, D.; Poon, V.K.; Chan, C.C.; Leung, H.C.; Fai, N.; Lin, Y.P.; et al. A novel peptide with potent and broad-spectrum antiviral activities against multiple respiratory viruses. Sci. Rep. 2016, 6, 22008. [Google Scholar] [CrossRef]
- Sample, C.J.; Hudak, K.E.; Barefoot, B.E.; Koci, M.D.; Wanyonyi, M.S.; Abraham, S.; Staats, H.F.; Ramsburg, E.A. A mastoparan-derived peptide has broad-spectrum antiviral activity against enveloped viruses. Peptides 2013, 48, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhao, Z.; Zhou, D.; Chen, Y.; Hong, W.; Cao, L.; Yang, J.; Zhang, Y.; Shi, W.; Cao, Z.; et al. Virucidal activity of a scorpion venom peptide variant mucroporin-M1 against measles, SARS-CoV and influenza H5N1 viruses. Peptides 2011, 32, 1518–1525. [Google Scholar] [CrossRef]
- Lu, S.; Pan, X.; Chen, D.; Xie, X.; Wu, Y.; Shang, W.; Jiang, X.; Sun, Y.; Fan, S.; He, J. Broad-spectrum antivirals of protoporphyrins inhibit the entry of highly pathogenic emerging viruses. Bioorg. Chem. 2021, 107, 104619. [Google Scholar] [CrossRef]
- ElSawy, K.M.; Twarock, R.; Verma, C.S.; Caves, L.S. Peptide inhibitors of viral assembly: A novel route to broad-spectrum antivirals. J. Chem. Inf. Model. 2012, 52, 770–776. [Google Scholar] [CrossRef]
- Brugger, B.; Glass, B.; Haberkant, P.; Leibrecht, I.; Wieland, F.T.; Krausslich, H.G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646. [Google Scholar] [CrossRef] [Green Version]
- Borkotoky, S.; Dey, D.; Banerjee, M. Computational Insight Into the Mechanism of SARS-CoV-2 Membrane Fusion. J. Chem. Inf. Model. 2021, 61, 423–431. [Google Scholar] [CrossRef]
- Meher, G.; Chakraborty, H. Membrane Composition Modulates Fusion by Altering Membrane Properties and Fusion Peptide Structure. J. Membr. Biol. 2019, 252, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Pattnaik, G.P.; Meher, G.; Chakraborty, H. Exploring the Mechanism of Viral Peptide-Induced Membrane Fusion. Adv. Exp. Med. Biol. 2018, 1112, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M. Fusion peptides and the mechanism of viral fusion. Biochim. Biophys. Acta 2003, 1614, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Pessi, A.; Bixler, S.L.; Soloveva, V.; Radoshitzky, S.; Retterer, C.; Kenny, T.; Zamani, R.; Gomba, G.; Gharabeih, D.; Wells, J.; et al. Cholesterol-conjugated stapled peptides inhibit Ebola and Marburg viruses in vitro and in vivo. Antivir. Res. 2019, 171, 104592. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.J.; Dvory-Sobol, H.; Xiong, A.; Cho, S.J.; Frank, C.W.; Glenn, J.S. Mechanism of an amphipathic alpha-helical peptide’s antiviral activity involves size-dependent virus particle lysis. ACS Chem. Biol. 2009, 4, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [Green Version]
- Pessi, A. Cholesterol-conjugated peptide antivirals: A path to a rapid response to emerging viral diseases. J. Pept. Sci. 2015, 21, 379–386. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.D.; Koellhoffer, J.F.; Chandran, K.; Lai, J.R. C-peptide inhibitors of Ebola virus glycoprotein-mediated cell entry: Effects of conjugation to cholesterol and side chain-side chain crosslinking. Bioorg. Med. Chem. Lett. 2013, 23, 5356–5360. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.K.; Pessi, A.; Gui, L.; Santoprete, A.; Talekar, A.; Moscona, A.; Porotto, M. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 2011, 286, 42141–42149. [Google Scholar] [CrossRef] [Green Version]
- Li, C.G.; Tang, W.; Chi, X.J.; Dong, Z.M.; Wang, X.X.; Wang, X.J. A cholesterol tag at the N terminus of the relatively broad-spectrum fusion inhibitory peptide targets an earlier stage of fusion glycoprotein activation and increases the peptide’s antiviral potency in vivo. J. Virol. 2013, 87, 9223–9232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, P.M.; Augusto, M.T.; Porotto, M.; Santos, N.C. The pH-sensitive action of cholesterol-conjugated peptide inhibitors of influenza virus. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183762. [Google Scholar] [CrossRef] [PubMed]
- Pessi, A.; Langella, A.; Capito, E.; Ghezzi, S.; Vicenzi, E.; Poli, G.; Ketas, T.; Mathieu, C.; Cortese, R.; Horvat, B.; et al. A general strategy to endow natural fusion-protein-derived peptides with potent antiviral activity. PLoS ONE 2012, 7, e36833. [Google Scholar] [CrossRef]
- Gomes, B.; Santos, N.C.; Porotto, M. Biophysical Properties and Antiviral Activities of Measles Fusion Protein Derived Peptide Conjugated with 25-Hydroxycholesterol. Molecules 2017, 22, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porotto, M.; Rockx, B.; Yokoyama, C.C.; Talekar, A.; Devito, I.; Palermo, L.M.; Liu, J.; Cortese, R.; Lu, M.; Feldmann, H.; et al. Inhibition of Nipah virus infection in vivo: Targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 2010, 6, e1001168. [Google Scholar] [CrossRef] [PubMed]
- Figueira, T.N.; Palermo, L.M.; Veiga, A.S.; Huey, D.; Alabi, C.A.; Santos, N.C.; Welsch, J.C.; Mathieu, C.; Horvat, B.; Niewiesk, S.; et al. In Vivo Efficacy of Measles Virus Fusion Protein-Derived Peptides Is Modulated by the Properties of Self-Assembly and Membrane Residence. J. Virol. 2017, 91, e01554-16. [Google Scholar] [CrossRef] [Green Version]
- Merlino, F.; Tomassi, S.; Yousif, A.M.; Messere, A.; Marinelli, L.; Grieco, P.; Novellino, E.; Cosconati, S.; Di Maro, S. Boosting Fmoc Solid-Phase Peptide Synthesis by Ultrasonication. Org. Lett. 2019, 21, 6378–6382. [Google Scholar] [CrossRef]
- Lombardi, L.; Falanga, A.; Del Genio, V.; Palomba, L.; Galdiero, M.; Franci, G.; Galdiero, S. A boost to the antiviral activity: Cholesterol tagged peptides derived from glycoprotein B of Herpes Simplex virus type I. Int. J. Biol. Macromol. 2020, 162, 882–893. [Google Scholar] [CrossRef]
- Bellavita, R.; Falanga, A.; Buommino, E.; Merlino, F.; Casciaro, B.; Cappiello, F.; Mangoni, M.L.; Novellino, E.; Catania, M.R.; Paolillo, R.; et al. Novel temporin L antimicrobial peptides: Promoting self-assembling by lipidic tags to tackle superbugs. J Enzyme Inhib. Med. Chem. 2020, 35, 1751–1764. [Google Scholar] [CrossRef]
- Ilyushina, N.A.; Ikizler, M.R.; Kawaoka, Y.; Rudenko, L.G.; Treanor, J.J.; Subbarao, K.; Wright, P.F. Comparative study of influenza virus replication in MDCK cells and in primary cells derived from adenoids and airway epithelium. J. Virol. 2012, 86, 11725–11734. [Google Scholar] [CrossRef] [Green Version]
- Cantisani, M.; Finamore, E.; Mignogna, E.; Falanga, A.; Nicoletti, G.F.; Pedone, C.; Morelli, G.; Leone, M.; Galdiero, M.; Galdiero, S. Structural insights into and activity analysis of the antimicrobial peptide myxinidin. Antimicrob. Agents Chemother. 2014, 58, 5280–5290. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Code | Sequence | MW (as TFA Salt) | %Helix in DPC * |
|---|---|---|---|---|
| Temporin L | TL | H-Phe-Val-Gln-Trp-Phe-Ser-Lys-Phe-Leu-Gly-Arg-Ile-Leu-NH2 | 1640.02 | 62.2 |
| [Pro3, DLeu9] TL | TL1 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Gly-Arg-Ile-Leu-NH2 | 1949.98 | 42 |
| Name | Code | Sequence | MW (as TFA Salt) | %Helix in DPC * |
|---|---|---|---|---|
| [Pro3,DLeu9] TL | TL1 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Gly-Arg-Ile-Leu-NH2 | 1949.98 | 42 |
| [Pro3,DLeu9,Pro10] TL | TL2 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Pro-Arg-Ile-Leu-NH2 | 1990.01 | 15 |
| [Pro3,DLeu9,DPro10] TL | TL3 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-DPro-Arg-Ile-Leu-NH2 | 1990.01 | 8 |
| [Pro3,DLeu9,Hyp10] TL | TL4 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Hyp-Arg-Ile-Leu-NH2 | 2006.01 | 18 |
| [Pro3,DLeu9,DHyp10] TL | TL5 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-DHyp-Arg-Ile-Leu-NH2 | 2006.01 | 9 |
| [Pro3,DLeu9,Nle10] TL | TL6 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-NH2 | 2006.04 | 51 |
| [Pro3,DLeu9,DNle10] TL | TL7 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-DNle-Arg-Ile-Leu-NH2 | 2006.04 | 30 |
| [Pro3,DLeu9,Lys10] TL | TL8 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Lys-Arg-Ile-Leu-NH2 | 2135.07 | 61 |
| [Pro3,DLeu9,DLys10] TL | TL9 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-DLys-Arg-Ile-Leu-NH2 | 2079.04 | 30 |
| [Pro3,DLeu9,Trp10] TL | TL10 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Trp-Arg-Ile-Leu-NH2 | 2079.04 | 50 |
| [Pro3,DLeu9,DTrp10] TL | TL11 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-DTrp-Arg-Ile-Leu-NH2 | 2079.04 | 40 |
| [Pro3,DLeu9,Aic10] TL | TL12 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Aic-Arg-Ile-Leu-NH2 | 2053.03 | 18 |
| CC50 | IC90 | IC50 | TI | ||||
|---|---|---|---|---|---|---|---|
| HSV-1 | SARS-CoV-2 | HSV-1 | SARS-CoV-2 | HSV-1 | SARS-CoV-2 | ||
| TL1 | 42.18 | 18.69 | 12.14 | 9.99 | 4.62 | 4.22 | 9.13 |
| TL2 | 56.52 | 18.76 | 13.98 | 7.70 | 4.82 | 7.34 | 11.73 |
| TL3 | >100.00 | >50.00 | >50.00 | >50.00 | >50.00 | - | - |
| TL4 | 60.19 | 15.89 | 25.00 | 7.73 | 6.72 | 7.88 | 9.06 |
| TL5 | >100.00 | >50.00 | >50.00 | >50.00 | >50.00 | - | - |
| TL6 | 64.52 | 9.12 | 12.15 | 2.66 | 0.53 | 24.26 | 121.74 |
| TL7 | 32.16 | 12.83 | 17.41 | 3.65 | 1.00 | 8.81 | 32.16 |
| TL8 | 22.32 | 10.84 | 12.93 | 2.49 | 0.88 | 8.96 | 25.36 |
| TL9 | 45.92 | 12.71 | 25.33 | 3.53 | 4.63 | 13.01 | 9.92 |
| TL10 | 35.47 | 8.25 | 27.71 | 3.01 | 0.88 | 11.78 | 40.31 |
| TL11 | 24.12 | 7.89 | 14.85 | 1.86 | 4.07 | 12.97 | 5.93 |
| TL12 | 8.28 | 1.58 | 13.85 | 0.68 | 0.65 | 12.18 | 12.74 |
| CC50 | IC90 | IC50 | TI | ||||
|---|---|---|---|---|---|---|---|
| MeV | Influenza | MeV | Influenza | MeV | Influenza | ||
| TL1 | 42.18 | >50.00 | 12.52 | >50.00 | 6.74 | - | 6.26 |
| TL2 | 56.52 | >50.00 | 21.28 | >50.00 | 11.95 | - | 4.73 |
| TL3 | >100.00 | >50.00 | >50.00 | >50.00 | >50.00 | - | - |
| TL4 | 60.90 | >50.00 | 31.65 | >50.00 | 16.26 | - | 3.75 |
| TL5 | >100.00 | >50.00 | >50.00 | >50.00 | >50.00 | - | - |
| TL6 | 64.52 | >50.00 | 9.34 | 34.58 | 2.66 | 1.87 | 24.26 |
| TL7 | 32.16 | >50.00 | 13.49 | >50.00 | 4.45 | - | 7.23 |
| TL8 | 22.32 | >50.00 | 9.78 | 37.10 | 3.40 | 0.60 | 6.56 |
| TL9 | 45.92 | >50.00 | 11.18 | 44.98 | 3.44 | 1.02 | 13.35 |
| TL10 | 35.47 | >50.00 | 12.09 | 42.82 | 3.79 | 0.83 | 9.36 |
| TL11 | 24.12 | >50.00 | 9.77 | 43.23 | 3.05 | 0.56 | 7.91 |
| TL12 | 8.28 | >50.00 | 13.88 | 30.42 | 0.71 | 0.27 | 11.66 |
| Name | Code | Sequence | MW (as TFA Salt) |
|---|---|---|---|
| [Pro3,DLeu9,Nle10] TL | TL6 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-NH2 | 2006.04 |
| [Pro3,DLeu9,Nle10] TL-C-CHOL | TL6.1 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-Cys-[PEG4-Cholesterol]-NH2 | 2708.10 |
| [Pro3,DLeu9,Nle10] TL-GGC-CHOL | TL6.2 | H-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-Gly-Gly-Cys-[PEG4-Cholesterol]-NH2 | 2858.24 |
| CHOL-C-[Pro3,DLeu9,Nle10] TL | TL6.3 | H-[Cholesterol-PEG4]-Cys-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-NH2 | 2708.10 |
| CHOL-CGG-[Pro3,DLeu9,Nle10] TL | TL6.4 | H-[Cholesterol-PEG4]-Cys-Gly-Gly-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-NH2 | 2858.24 |
| Undecanoic-[Pro3,DLeu9,Nle10] TL | TL6.5 | H-Undecanoic acid-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-NH2 | 2192.33 |
| Tridecanoic-[Pro3,DLeu9,Nle10] TL | TL6.6 | H-Tridecanoic acid-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-NH2 | 2220.39 |
| Pentadecanoic-[Pro3,DLeu9,Nle10] TL | TL6.7 | H-Pentadecanoic acid-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-NH2 | 2248.44 |
| Hexadecenoic-[Pro3,DLeu9,Nle10] TL | TL6.8 | H-Palmitic acid-Phe-Val-Pro-Trp-Phe-Ser-Lys-Phe-DLeu-Nle-Arg-Ile-Leu-NH2 | 2262.47 |
| CC50 | IC90 | IC50 | TI | ||||
|---|---|---|---|---|---|---|---|
| HSV-1 | SARS-CoV-2 | HSV-1 | SARS-CoV-2 | HSV-1 | SARS-CoV-2 | ||
| TL6 | 64.5 | 9.12 | 12.15 | 2.66 | 0.53 | 24.25 | 121.70 |
| TL6.1 | >100.00 | 11.13 | 12.03 | 6.4 | 3.18 | 15.63 | 31.45 |
| TL6.2 | >100.00 | 47.95 | 49.95 | 9.54 | 12.12 | 10.48 | 8.25 |
| TL6.3 | >100.00 | 2.19 | 1.89 | 0.89 | 0.76 | 112.36 | 131.58 |
| TL6.4 | >100.00 | 8.21 | 10.21 | 2.32 | 1.02 | 43.10 | 98.04 |
| TL6.5 | >100.00 | <0.10 | <0.10 | <0.10 | <0.10 | 1000.00 | 1000.00 |
| TL6.6 | >100.00 | 2.86 | 5.86 | 1.39 | 0.39 | 71.94 | 256.41 |
| TL6.7 | >100.00 | 4.86 | 7.86 | 2.48 | 0.48 | 40.32 | 208.33 |
| TL6.8 | >100.00 | 4.96 | 6.96 | 3.13 | 0.77 | 31.95 | 129.87 |
| CC50 | IC90 | IC50 | TI | ||||
|---|---|---|---|---|---|---|---|
| MeV | Influenza | MeV | Influenza | MeV | Influenza | ||
| TL6 | 64.52 | >50.00 | 9.12 | 34.58 | 2.66 | 1.87 | 24.26 |
| TL6.1 | >100.00 | >50.00 | 10.02 | 39.1 | 5.55 | 2.56 | 18.02 |
| TL6.2 | >100.00 | >50.00 | 49.90 | 40.89 | 21.12 | 2.45 | 4.73 |
| TL6.3 | >100.00 | 31.10 | 5.89 | 22.3 | 2.55 | 4.48 | 39.22 |
| TL6.4 | >100.00 | 49.90 | 9.90 | 28.88 | 6.02 | 3.46 | 16.61 |
| TL6.5 | >100.00 | 29.18 | <0.10 | 10.01 | <0.10 | 1000.00 | 1000.00 |
| TL6.6 | >100.00 | >50.00 | 6.66 | 33.36 | 3.39 | 3.00 | 29.50 |
| TL6.7 | >100.00 | >50.00 | 8.10 | 37.77 | 3.48 | 2.65 | 28.74 |
| TL6.8 | >100.00 | >50.00 | 7.11 | 39.4 | 4.77 | 2.54 | 20.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zannella, C.; Chianese, A.; Palomba, L.; Marcocci, M.E.; Bellavita, R.; Merlino, F.; Grieco, P.; Folliero, V.; De Filippis, A.; Mangoni, M.; et al. Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs. Int. J. Mol. Sci. 2022, 23, 2060. https://doi.org/10.3390/ijms23042060
Zannella C, Chianese A, Palomba L, Marcocci ME, Bellavita R, Merlino F, Grieco P, Folliero V, De Filippis A, Mangoni M, et al. Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs. International Journal of Molecular Sciences. 2022; 23(4):2060. https://doi.org/10.3390/ijms23042060
Chicago/Turabian StyleZannella, Carla, Annalisa Chianese, Luciana Palomba, Maria Elena Marcocci, Rosa Bellavita, Francesco Merlino, Paolo Grieco, Veronica Folliero, Anna De Filippis, Marialuisa Mangoni, and et al. 2022. "Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs" International Journal of Molecular Sciences 23, no. 4: 2060. https://doi.org/10.3390/ijms23042060