
Metastasising Fibroblasts Show an HDAC6-Dependent Increase in Migration Speed and Loss of Directionality Linked to Major Changes in the Vimentin Interactome

 , , , ,
, , , ,
Abstract
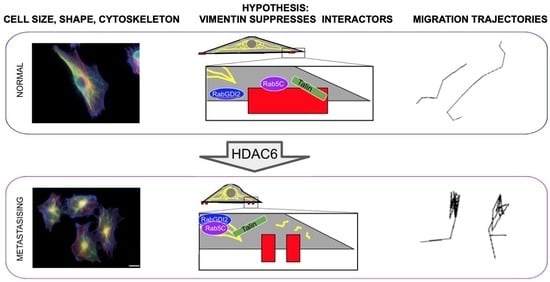
:
1. Introduction
2. Results
2.1. SV40T and H-RasV12 Change the Spatial Organisation of Vimentin More Than That of Actin and Tubulin
2.2. HDAC6 Is Required for the Shape and Motility of Metastasising Cells
2.3. Inhibition of HDAC6 Activity Results in Loss of Nanoscale, Non-Filamentous, Short Versions of Vimentin at the Cell Periphery, and Formation of Cage-like Clusters/Asters of Filamentous Vimentin
2.4. H-RasV12 and HDAC6 Activities Change the Binding of Cytoskeletal, Cell–Extracellular Matrix Adhesion Components to Vimentin Intermediate Filaments
3. Discussion
4. Materials and Methods
4.1. Cell Clture and Treatments
4.2. Immunofluorescence Staining, Confocal and STED Imaging and Computational Analysis of STED Images
4.3. Live-Cell Imaging
4.4. Invasion Assay
4.5. Image Analysis
4.6. Statistical Analysis
4.7. Preparation of Enriched Intermediate Filament Protein Fraction
4.8. Western Blotting
4.9. Quantitative Proteomic, Mass Spectrometry-Based Analysis of the Vimentin Interactome
4.10. Protein Identification, Relative Quantification, Bioinformatic Functional Profiling and Interaction Analysis
4.11. Tracking and Analysis of Migrating Cells
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eckes, B.; Dogic, D.; Colucci-Guyon, E.; Wang, N.; Maniotis, A.; Ingber, D.; Merckling, A.; Langa, F.; Aumailley, M.; Delouvée, A.; et al. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 1998, 111, 1897–1907. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Trevor, K.T. Experimental co-expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am. J. Pathol. 1997, 150, 483–495. [Google Scholar] [PubMed]
- Rathje, L.-S.Z.; Nordgren, N.; Pettersson, T.; Ronnlund, D.; Widengren, J.; Aspenstrom, P.; Gad, A.K.B. Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness. Proc. Natl. Acad. Sci. USA 2014, 111, 1515–1520. [Google Scholar] [CrossRef] [Green Version]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.-P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2010, 30, 1436–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Xu, G.; Wu, M.; Zhang, Y.; Li, Q.; Liu, P.; Zhu, T.; Song, A.; Zhao, L.; Han, Z.; et al. Overexpression of vimentin contributes to prostate cancer invasion and metastasis via src regulation. Anticancer. Res. 2008, 28, 327–334. [Google Scholar]
- Zhu, Q.-S.; Rosenblatt, K.; Huang, K.-L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2010, 30, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfand, B.T.; Mendez, M.G.; Murthy, S.N.P.; Shumaker, D.K.; Grin, B.; Mahammad, S.; Aebi, U.; Wedig, T.; Wu, Y.I.; Hahn, K.M.; et al. Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 2011, 22, 1274–1289. [Google Scholar] [CrossRef]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell-Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2017, 24, 420–432. [Google Scholar] [CrossRef] [Green Version]
- Van Bodegraven, E.J.; Etienne-Manneville, S. Intermediate filaments against actomyosin: The David and Goliath of cell migration. Curr. Opin Cell Biol. 2020, 66, 79–88. [Google Scholar] [CrossRef]
- Battaglia, R.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1796. [Google Scholar] [CrossRef] [Green Version]
- Terriac, E.; Coceano, G.; Mavajian, Z.; Hageman, T.A.G.; Christ, A.F.; Testa, I.; Lautenschläger, F.; Gad, A.K.B. Vimentin Levels and Serine 71 Phosphorylation in the Control of Cell-Matrix Adhesions, Migration Speed, and Shape of Transformed Human Fibroblasts. Cells 2017, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Ostrowska-Podhorodecka, Z.; Ding, I.; Lee, W.; Tanic, J.; Abbasi, S.; Arora, P.D.; Liu, R.S.; Patteson, A.E.; Janmey, P.A.; McCulloch, C.A. Vimentin tunes cell migration on collagen by controlling beta1 integrin activation and clustering. J. Cell Sci. 2021, 134, jcs254359. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lin, H.H.; Tang, M.J.; Wang, Y.K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [Green Version]
- Lynch, C.D.; Lazar, A.M.; Iskratsch, T.; Zhang, X.; Sheetz, M.P. Endoplasmic spreading requires coalescence of vimentin intermediate filaments at force-bearing adhesions. Mol. Biol. Cell 2013, 24, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Bismar, T.A.; Su, J.; Xu, B.; Kristiansen, G.; Varga, Z.; Teng, L.; Ingber, D.E.; Mammoto, A.; Kumar, R.; et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J. Exp. Med. 2010, 207, 2421–2437. [Google Scholar] [CrossRef] [Green Version]
- Challa, A.A.; Stefanovic, B. A Novel Role of Vimentin Filaments: Binding and Stabilization of Collagen mRNAs. Mol. Cell. Biol. 2011, 31, 3773–3789. [Google Scholar] [CrossRef] [Green Version]
- Challa, A.A.; Vukmirovic, M.; Blackmon, J.; Stefanovic, B. Withaferin-A Reduces Type I Collagen Expression In Vitro and Inhibits Development of Myocardial Fibrosis In Vivo. PLoS ONE 2012, 7, e42989. [Google Scholar] [CrossRef] [PubMed]
- Rönnlund, D.; Gad, A.K.B.; Blom, H.; Aspenström, P.; Widengren, J. Spatial organization of proteins in metastasizing cells. Cytom. Part A 2013, 83, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-H.; Ma, Q.; Wu, H.-P.; Khamis, M.Y.; Li, Y.-H.; Ma, L.-Y.; Liu, H.-M. A Review of Progress in Histone Deacetylase 6 Inhibitors Research: Structural Specificity and Functional Diversity. J. Med. Chem. 2021, 64, 1362–1391. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, P.; Rupprecht, J.-F.; Wieser, S.; Ruprecht, V.; Bénichou, O.; Carpi, N.; Coppey, M.; De Beco, S.; Gov, N.; Heisenberg, C.-P.; et al. Actin Flows Mediate a Universal Coupling between Cell Speed and Cell Persistence. Cell 2015, 161, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Chen, M.; Hawks, C.L.; Pereira-Smith, O.M.; Hornsby, P.J. The Minimal Set of Genetic Alterations Required for Conversion of Primary Human Fibroblasts to Cancer Cells in the Subrenal Capsule Assay. Neoplasia 2005, 7, 585–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsson, F.; Skogs, M.; Huss, M.; Rexhepaj, E.; O’Hurley, G.; Klevebring, D.; Pontén, F.; Gad, A.; Uhlen, M.; Lundberg, E. Majority of differentially expressed genes are down-regulated during malignant transformation in a four-stage model. Proc. Natl. Acad. Sci. USA 2013, 110, 6853–6858. [Google Scholar] [CrossRef] [Green Version]
- Gan, Z.; Ding, L.; Burckhardt, C.J.; Lowery, J.; Zaritsky, A.; Sitterley, K.; Mota, A.; Costigliola, N.; Starker, C.; Voytas, D.; et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst. 2016, 3, 500–501. [Google Scholar] [CrossRef] [Green Version]
- Goldman, R.D. The role of three cytoplasmic fibers in bhk-21 cell motility. J. Cell Biol. 1971, 51, 752–762. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindström, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-beta-Slug signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef] [Green Version]
- Sanghvi-Shah, R.; Weber, G.F. Intermediate Filaments at the Junction of Mechanotransduction, Migration, and Development. Front. Cell Dev. Biol. 2017, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Angrand, P.-O.; Segura, I.; Völkel, P.; Ghidelli, S.; Terry, R.; Brajenovic, M.; Vintersten, K.; Klein, R.; Superti-Furga, G.; Drewes, G.; et al. Transgenic Mouse Proteomics Identifies New 14-3-3-associated Proteins Involved in Cytoskeletal Rearrangements and Cell Signaling. Mol. Cell. Proteom. 2006, 5, 2211–2227. [Google Scholar] [CrossRef] [Green Version]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Chen, P.-I.; Schauer, K.; Kong, C.; Harding, A.R.; Goud, B.; Stahl, P.D. Rab5 Isoforms Orchestrate a “Division of Labor” in the Endocytic Network; Rab5C Modulates Rac-Mediated Cell Motility. PLoS ONE 2014, 9, e90384. [Google Scholar] [CrossRef] [Green Version]
- Velez-Delvalle, C.; Marsch-Moreno, M.; Castro-Muñozledo, F.; Galván-Mendoza, I.J.; Kuri-Harcuch, W. Epithelial cell migration requires the interaction between the vimentin and keratin intermediate filaments. Sci. Rep. 2016, 6, 24389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, S.J.; Rodgers, M.A.; Perrin, B.J.; Han, J.; Bennin, D.A.; Critchley, D.R.; Huttenlocher, A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 2004, 6, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, P.A.; Silva, P.; Díaz, J.; Arriagada, C.; Canales, J.; Cerda, O.; Torres, V.A. Calpain2 mediates Rab5-driven focal adhesion disassembly and cell migration. Cell Adhes. Migr. 2017, 12, 185–194. [Google Scholar] [CrossRef]
- Xu, L.; Braun, L.J.; Rönnlund, D.; Widengren, J.; Aspenström, P.; Gad, A.K.B. Nanoscale localization of proteins within focal adhesions indicates discrete functional assemblies with selective force-dependence. FEBS J. 2018, 285, 1635–1652. [Google Scholar] [CrossRef] [Green Version]
- Gad, A.; Lach, S.; Crimaldi, L.; Gimona, M. Plectin deposition at podosome rings requires myosin contractility. Cell Motil. Cytoskelet. 2008, 65, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.S.; Porter, T.J.; Xu, N.; Arndt, Z.P.; Ako-Asare, K.; Heo, H.; Thompson, E.A.; Moore, D.L. Vimentin Coordinates Protein Turnover at the Aggresome during Neural Stem Cell Quiescence Exit. Cell Stem Cell 2020, 26, 558–568.e9. [Google Scholar] [CrossRef] [PubMed]
- Shaebani, M.R.; Jose, R.; Santen, L.; Stankevicins, L.; Lautenschläger, F. Persistence-Speed Coupling Enhances the Search Efficiency of Migrating Immune Cells. Phys. Rev. Lett. 2020, 125, 268102. [Google Scholar] [CrossRef]
- Gad, A.; Thullberg, M.; Dannenberg, J.H.; te Riele, H.; Strömblad, S. Retinoblastoma susceptibility gene product (pRb) and p107 functionally separate the requirements for serum and an-chorage in the cell cycle G1-phase. J. Biol. Chem. 2004, 279, 13640–13644. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, H.; Kreplak, L.; Aebi, U. Isolation, Characterization, and In Vitro Assembly of Intermediate Filaments. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2004; Volume 78, pp. 3–24. [Google Scholar] [CrossRef]
- Leech, S.H.; Evans, C.A.; Shaw, L.; Wong, C.H.; Connolly, J.; Griffiths, J.R.; Whetton, A.D.; Corfe, B.M. Proteomic analyses of intermediate filaments reveals cytokeratin8 is highly acetylated—Implications for colorectal epithelial homeostasis. Proteomics 2008, 8, 279–288. [Google Scholar] [CrossRef]
- Mackinder, M.A.; Evans, C.A.; Chowdry, J.; Staton, C.A.; Corfe, B.M. Alteration in composition of keratin intermediate filaments in a model of breast cancer progression and the potential to reverse hallmarks of metastasis. Cancer Biomark. 2013, 12, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Alexeyenko, A.; Hennig, K.; Danielsson, F.; Lebbink, R.J.; Fielden, M.; Turunen, S.P.; Lehti, K.; Kashuba, V.; Madapura, H.S.; et al. RhoA knockout fibroblasts lose tumor-inhibitory capacity in vitro and promote tumor growth in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E1413–E1421. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteo-me-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342, Erratum in Nature 2013, 495, 126–127. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.D.; Goode, R.J.A.; Huang, C.; Powell, D.R.; Schittenhelm, R.B. LFQ-Analyst: An Easy-To-Use Interactive Web Platform to Analyze and Visualize Label-Free Proteomics Data Preprocessed with MaxQuant. J. Proteome Res. 2019, 19, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Tinevez, J.-Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene | Protein ID | BjhtertSV40TRasV12 vs. Bjhtert (Fold-Change) a |
|---|---|---|---|
| Protein SON | SON | P18583 | 5.5 |
| Neuroblast differentiation-associated protein AHNAK | AHNAK | Q09666 | 4.7 |
| WD repeat-containing protein 46 | WDR46 | O15213 | 4.5 |
| 60S ribosomal protein L7-like 1 | RPL7L1 | Q6DKI1 | 4.2 |
| Ribosome biogenesis protein BOP1 | BOP1 | Q14137 | 4.1 |
| RNA-binding protein 8A | RBM8A | Q9Y5S9 | 4.0 |
| Transcription factor BTF3 | BTF3 | A0A7I2YQL2 | 3.7 |
| Triosephosphate isomerase | TPI1 | P60174 | 3.7 |
| Elongation factor 1-β | EEF1B2 | P24534 | 3.6 |
| Keratin, type I cytoskeletal 18 | KRT18 | P05783 | 3.5 |
| U3 small nucleolar RNA-associated protein 15 homologue | UTP15 | Q8TED0 | 3.2 |
| WD repeat-containing protein 75 | WDR75 | Q8IWA0 | 3.2 |
| Guanine nucleotide-binding protein-like 3 | GNL3 | Q9BVP2 | 3.1 |
| Nuclear pore complex protein Nup153 | NUP153 | P49790 | 3.1 |
| DNA-directed RNA polymerase II subunit RPB3 | POLR2C | P19387 | 3.0 |
| U3 small nucleolar ribonucleoprotein protein MPP10 | MPHOSPH10 | O00566 | 2.9 |
| ATP-dependent RNA helicase DDX18 | DDX18 | Q9NVP1 | 2.9 |
| Deoxynucleotidyltransferase terminal-interacting protein 2 | DNTTIP2 | Q5QJE6 | 2.8 |
| Ribosome production factor 2 homologue | RPF2 | Q9H7B2 | 2.6 |
| Ras-related protein Rab5C | RAB5C | P51148 | 2.6 |
| H/ACA ribonucleoprotein complex subunit 4 | DKC1 | O60832 | 2.5 |
| MKI67 FHA-domain-interacting nucleolar phosphoprotein | NIFK | Q9BYG3 | 2.4 |
| Ribosome biogenesis protein BMS1 homologue | BMS1 | Q14692 | 2.2 |
| Nucleolar GTP-binding protein 1 | GTPBP4 | Q9BZE4 | 2.2 |
| Voltage-dependent anion-selective channel protein 2 | VDAC2 | P45880 | −2.1 |
| Histone H2B type 2-E | HIST2H2BE | Q16778 | −2.1 |
| RuvB-like 1 | RUVBL1 | Q9Y265 | −2.2 |
| Actin, α skeletal muscle | ACTA1 | P68133 | −2.2 |
| Bcl-2-associated transcription factor 1 | BCLAF1 | A0A1W2PQ43 | −2.2 |
| Testis-expressed sequence 10 protein | TEX10 | Q9NXF1 | −2.5 |
| HLA class I histocompatibility antigen, Cw-7 α chain | HLA-C.1 | O19617 | −2.5 |
| CD109 antigen | CD109 | Q6YHK3 | −2.6 |
| Collagen α-1(XII) chain | COL12A1 | D6RGG3 | −2.6 |
| HLA class I histocompatibility antigen, A-3 α chain | HLA-A | Q5SUL5 | −2.6 |
| Procollagen-lysine,2-oxoglutarate 5-dioxygenase 1/lysyl hydroxylase | PLOD1 | Q02809 | −2.6 |
| Heterogeneous nuclear ribonucleoprotein H3 | HNRNPH3 | P31942 | −2.8 |
| Lamina-associated polypeptide 2, isoform α/ thymopoietin | TMPO | P42166 | −2.9 |
| WD repeat-containing protein 18 | WDR18 | U3KQC1 | −2.9 |
| Heterogeneous nuclear ribonucleoprotein U-like protein 2 | HNRNPUL2-BSCL2 | H3BQZ7 | −3.1 |
| HLA class I histocompatibility antigen B α chain | HLA-B | Q2L6G2 | −3.2 |
| Voltage-dependent anion-selective channel protein 1 | VDAC1 | P21796 | −3.2 |
| Fibulin-2 | FBLN2 | P98095 | −3.5 |
| Basement membrane-specific heparan sulphate proteoglycan core protein | HSPG2 | P98160 | −4.1 |
| Interferon-induced GTP-binding protein Mx1 | MX1 | A0A7P0T9R0 | −4.4 |
| 26S proteasome non-ATPase regulatory subunit 6 | PSMD6 | Q15008 | −4.6 |
| Myosin regulatory light chain 12A | MYL12A | J3QRS3 | −6.8 |
| Interferon-induced GTP-binding protein Mx2 | MX2 | P20592 | −8.9 |
| Protein | Gene | Protein ID | BjhtertSv40TRasV12 with vs. without Tubacin (Fold-Change) a |
|---|---|---|---|
| 40S ribosomal protein S23 | RPS23 | P62266 | 2.1 |
| GTP-binding protein 4 | GTPBP4 | Q9BZE4 | 2.1 |
| 40S ribosomal protein S9 | RPS9 | P46781 | 2.2 |
| Probable 28S rRNA (cytosine(4447)-C(5))-methyltransferase | NOP2 | P46087 | 2.3 |
| mRNA turnover protein 4 homologue | MRTO4 | Q9UKD2 | 2.5 |
| Probable ATP-dependent RNA helicase DDX56 | DDX56 | G3V0G3 | 2.7 |
| U3 small nucleolar RNA-associated protein 6 homologue | UTP6 | Q9NYH9 | 3.1 |
| Periodic tryptophan protein 2 homologue | PWP2 | Q15269 | 4.3 |
| KRR1 small subunit processome component homologue | KRR1 | Q13601 | 4.9 |
| 26S protease regulatory subunit 10B | PSMC6 | A0A087X2I1 | −2.0 |
| Glycine--tRNA ligase | GARS | A0A6Q8PGW4 | −2.1 |
| Transitional endoplasmic reticulum ATPase | VCP | P55072 | −2.1 |
| X-ray repair cross-complementing protein 6 | XRCC6 | P12956 | −2.1 |
| Eukaryotic translation initiation factor 5B | EIF5B | A0A087WUT6 | −2.1 |
| T-complex protein 1 subunit γ | CCT3 | B4DUR8 | −2.1 |
| Emilin-1 | EMILIN1 | Q9Y6C2 | −2.3 |
| Glucose-6-phosphate 1-dehydrogenase | G6PD | P11413 | −2.4 |
| Prohibitin | PHB | P35232 | −2.5 |
| Rab GDP dissociation inhibitor β | GDI2 | P50395 | −2.5 |
| Tubulin-specific chaperone A | TBCA | E5RIW3 | −2.5 |
| Dolichyl-diphospho-oligosaccharide--protein glycosyltransferase 48 kDa subunit | DDOST | A0A0C4DGS1 | −2.6 |
| 26S proteasome non-ATPase regulatory subunit 2 | PSMD2 | Q13200 | −2.6 |
| Malate dehydrogenase, mitochondrial | MDH2 | P40926 | −2.7 |
| mRNA export factor | RAE1 | E9PQ57 | −2.7 |
| Coatomer subunit γ-1 | COPG1 | Q9Y678 | −2.8 |
| Voltage-dependent anion-selective channel protein 2/voltage dependent anion channel 2 | VDAC2 | P45880 | −2.9 |
| Heterogeneous nuclear ribonucleoprotein R | HNRNPR | O43390 | −2.9 |
| Heat shock protein β-1 | HSPB1 | P04792 | −3.0 |
| Elongation factor 1-β | EEF1B2 | P24534 | −3.1 |
| Neutral α-glucosidase AB | GANAB | Q14697 | −3.1 |
| Talin-1 | TLN1 | Q9Y490 | −3.1 |
| Peroxiredoxin-6 | PRDX6 | P30041 | −3.2 |
| Sodium/potassium-transporting ATPase subunit α-1 | ATP1A1 | P05023 | −3.2 |
| HLA class I histocompatibility antigen B α chain | HLA-B | Q2L6G2 | −3.3 |
| Flotillin-1 | FLOT1 | O75955 | −3.4 |
| Leucine-rich repeat-containing protein 59 | LRRC59 | Q96AG4 | −3.4 |
| tRNA-splicing ligase RtcB homologue | RTCB | Q9Y3I0 | −3.4 |
| Transcription intermediary factor 1-β | TRIM28 | Q13263 | −3.5 |
| Serpin H1 | SERPINH1 | P50454 | −3.6 |
| Interferon-induced GTP-binding protein Mx1/dynamin like GTPase 1 | MX1 | A0A7P0T9R0 | −3.6 |
| Signal transducer and activator of transcription 1-α/β | STAT1 | A0A669KB68 | −3.8 |
| Protein CutA | CUTA | O60888 | −3.9 |
| Protein disulphide-isomerase/collagen prolyl 4-hydroxylase β | P4HB | A0A7P0T8J3 | −4.3 |
| Basement-membrane-specific heparan sulphate proteoglycan core protein | HSPG2 | P98160 | −4.6 |
| KH domain-containing, RNA-binding, signal transduction-associated protein 1 | KHDRBS1 | Q07666 | −4.6 |
| Collagen α-1(XII) chain | COL12A1 | D6RGG3 | −4.8 |
| Tropomyosin α-4 chain | TPM4 | P67936 | −4.8 |
| Voltage-dependent anion-selective channel protein 1 | VDAC1 | P21796 | −4.9 |
| Prothymosin α | PTMA | B8ZZQ6 | −5.0 |
| Protein S100-A11 | S100A11 | P31949 | −5.4 |
| C-1-tetrahydrofolate synthase, cytoplasmic | MTHFD1 | V9GYY3 | −5.7 |
| HLA class I histocompatibility antigen, A-3 α chain | HLA-A | Q5SUL5 | −11.6 |
| RPLP1 | RPLP1 | P05386 | −50.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, C.A.; Kim, H.R.; Macfarlane, S.C.; Nowicki, P.I.A.; Baltes, C.; Xu, L.; Widengren, J.; Lautenschläger, F.; Corfe, B.M.; Gad, A.K.B. Metastasising Fibroblasts Show an HDAC6-Dependent Increase in Migration Speed and Loss of Directionality Linked to Major Changes in the Vimentin Interactome. Int. J. Mol. Sci. 2022, 23, 1961. https://doi.org/10.3390/ijms23041961
Evans CA, Kim HR, Macfarlane SC, Nowicki PIA, Baltes C, Xu L, Widengren J, Lautenschläger F, Corfe BM, Gad AKB. Metastasising Fibroblasts Show an HDAC6-Dependent Increase in Migration Speed and Loss of Directionality Linked to Major Changes in the Vimentin Interactome. International Journal of Molecular Sciences. 2022; 23(4):1961. https://doi.org/10.3390/ijms23041961
Chicago/Turabian StyleEvans, Caroline A., Hyejeong Rosemary Kim, Sarah C. Macfarlane, Poppy I. A. Nowicki, Carsten Baltes, Lei Xu, Jerker Widengren, Franziska Lautenschläger, Bernard M. Corfe, and Annica K. B. Gad. 2022. "Metastasising Fibroblasts Show an HDAC6-Dependent Increase in Migration Speed and Loss of Directionality Linked to Major Changes in the Vimentin Interactome" International Journal of Molecular Sciences 23, no. 4: 1961. https://doi.org/10.3390/ijms23041961