
The Role of Nutrients in Maintaining Hematopoietic Stem Cells and Healthy Hematopoiesis for Life

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
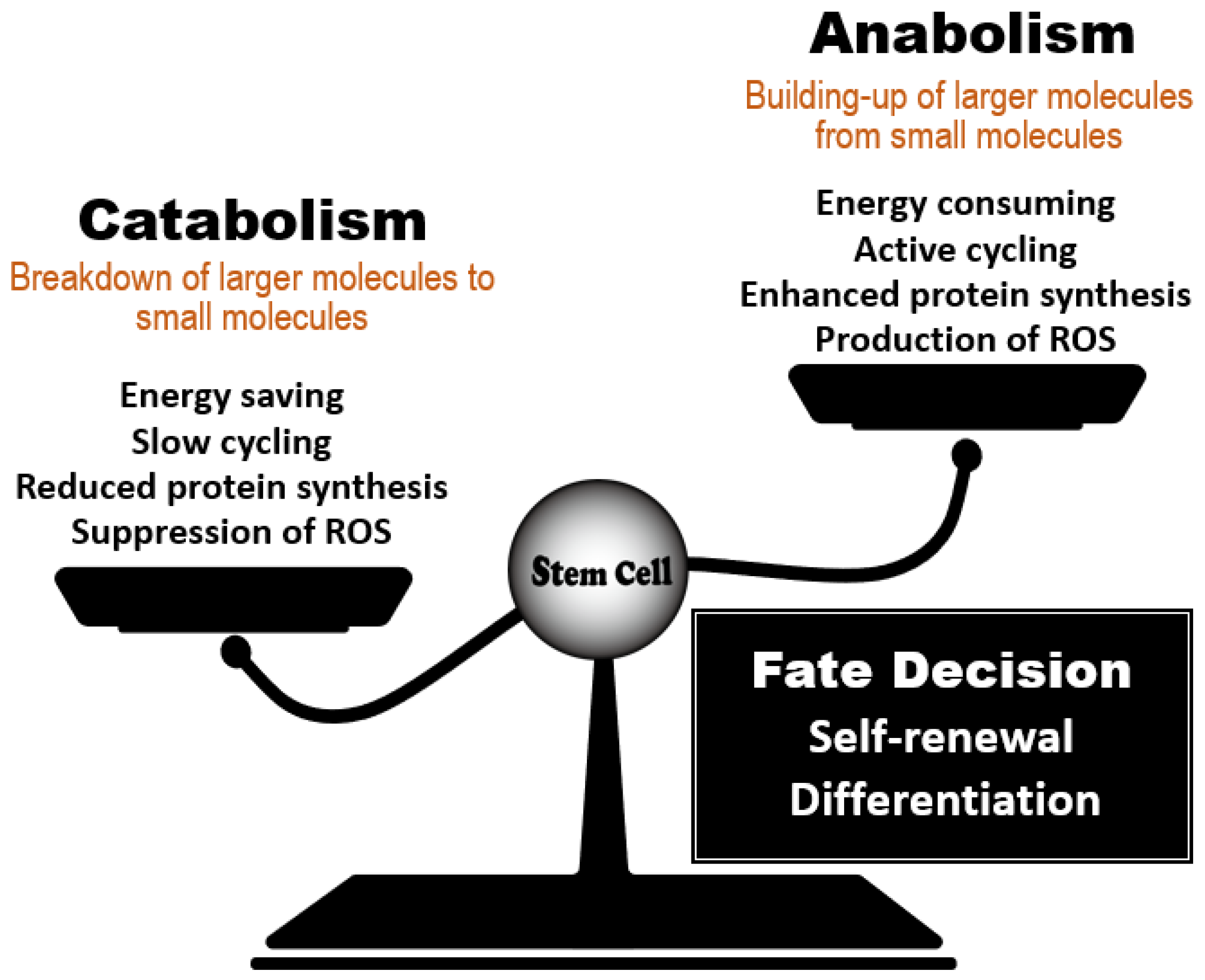
2. Effect of the Anabolic/Catabolic Balance on Stem Cell Homeostasis
2.1. Commonality of Nutrient-Sensing Regulators
2.2. Catabolic Regulators That Maintain Stem Cell Quiescence
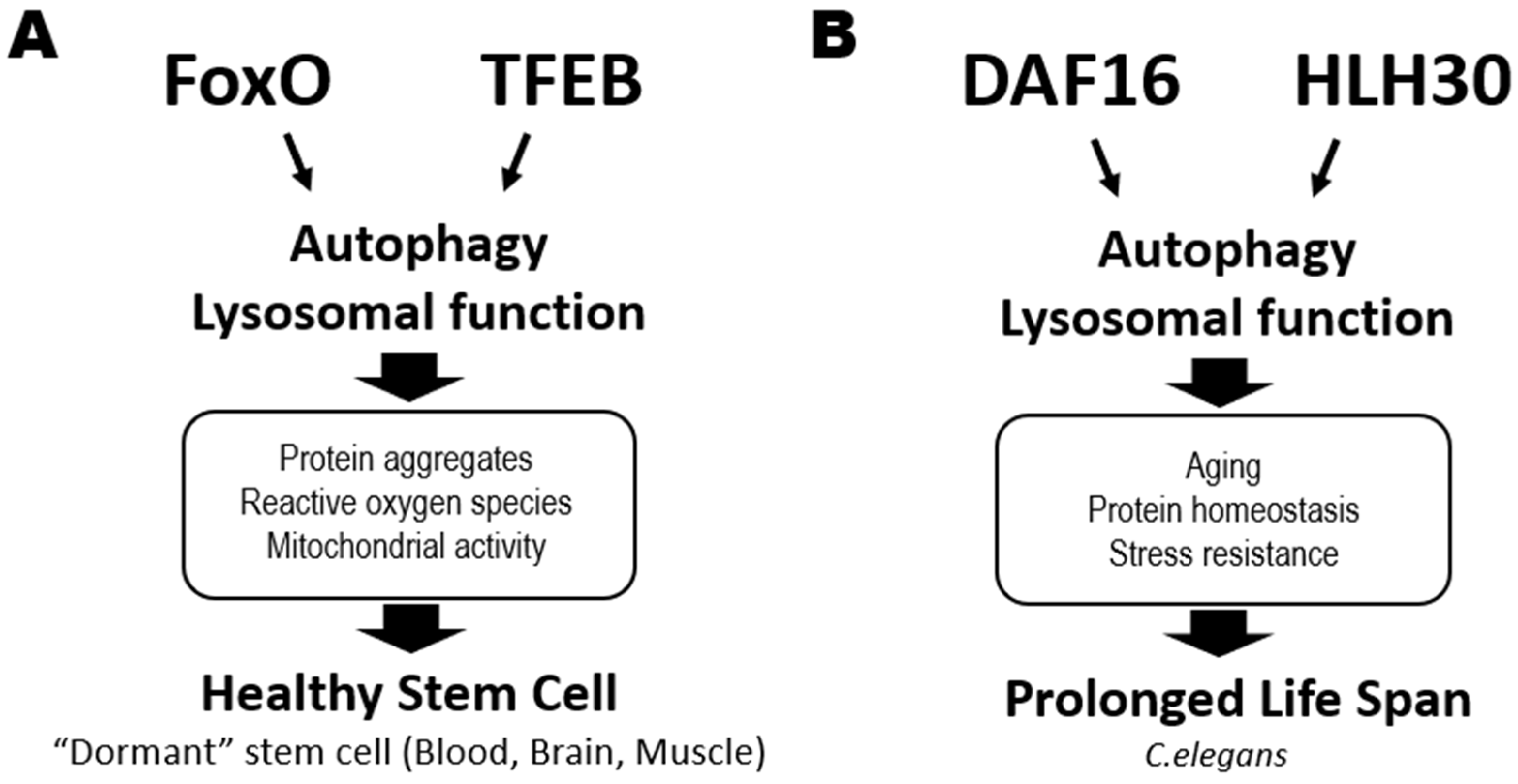
2.2.1. Autophagy Regulators
2.2.2. Transcription Factor EB (TFEB)
2.2.3. FoxO Family
2.2.4. Anabolic Regulators That Control HSC Self-Renewal
3. The Effects of Dietary Interventions on Stem Cell Homeostasis
3.1. Beneficial Effects of Dietary Restriction and Prolonged Fasting on HSC Functions
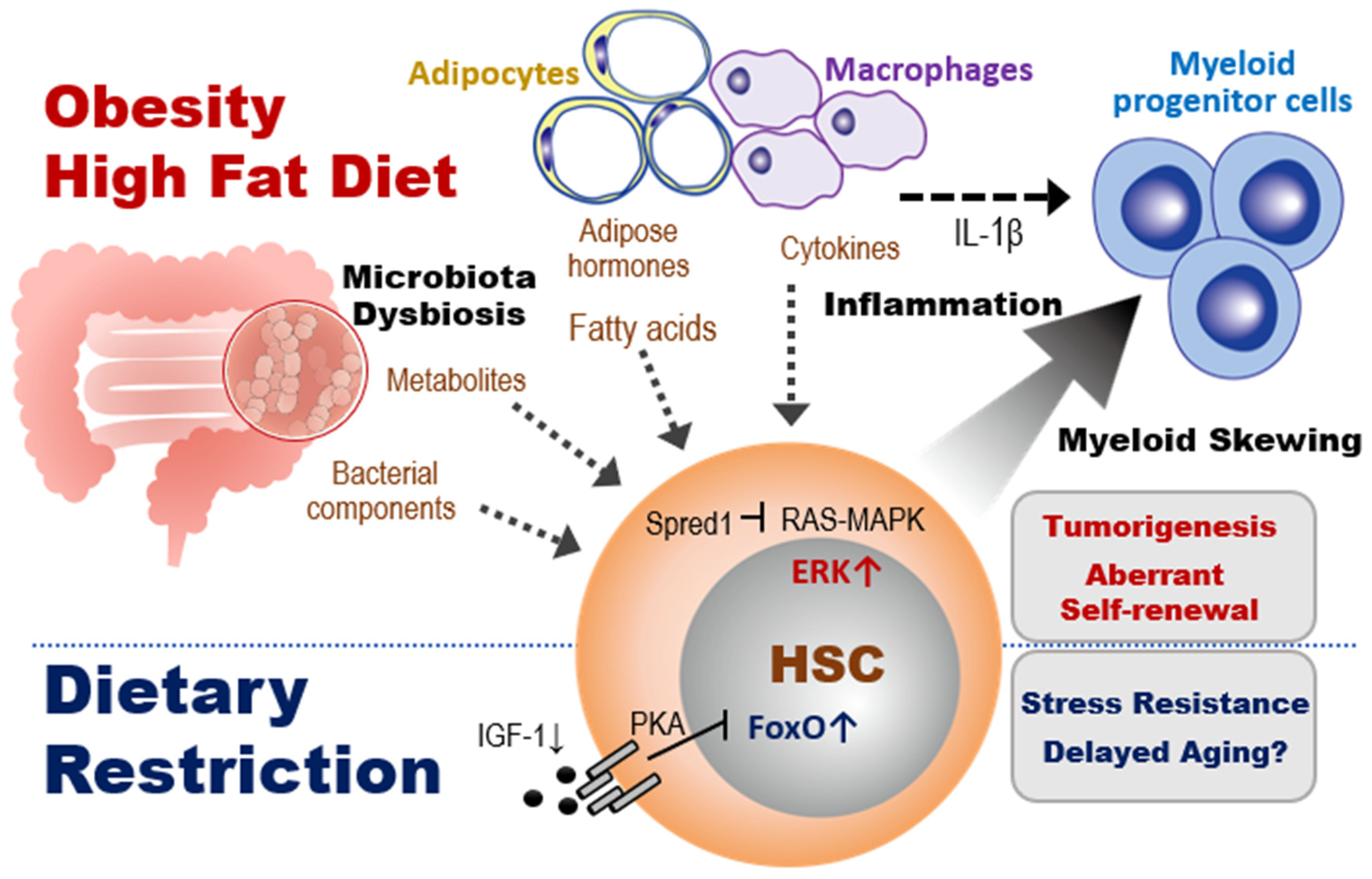
3.2. The Effects of Obesity on HSC Homeostasis
3.3. The Effects of HFD on HSC Homeostasis
3.4. The Effects of Metabolites Induced by Dietary Interventions on Stem Cell Fate Determination
3.4.1. Fatty Acid Composition
3.4.2. Ketone Bodies and Related Metabolites
3.4.3. Microbiota Producing Metabolites
4. Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nakamura-Ishizu, A.; Ito, K.; Suda, T. Hematopoietic Stem Cell Metabolism during Development and Aging. Dev. Cell 2020, 54, 239–255. [Google Scholar] [CrossRef]
- van Velthoven, C.T.J.; Rando, T.A. Stem Cell Quiescence: Dynamism, Restraint, and Cellular Idling. Cell Stem Cell 2019, 24, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Yoshimori, T. Autophagy and Longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar] [CrossRef]
- Novak, J.S.; Baksh, S.C.; Fuchs, E. Dietary interventions as regulators of stem cell behavior in homeostasis and disease. Genes Dev. 2021, 35, 199–211. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Dellavalle, A.; Diaz-Manera, J.; Messina, G.; Cossu, G. Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J. Clin. Investig. 2010, 120, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and Functional Implications of Adult Neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Mortensen, M.; Ferguson, D.; Edelmann, M.; Kessler, B.; Morten, K.J.; Komatsu, M.; Simon, A.K. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.J.; Williams, O.; de Boer, J.; Cain, K.C.; Macfarlane, M.; McGouran, J.; Kessler, B.M.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef]
- Jung, H.E.; Shim, Y.R.; Oh, J.E.; Oh, D.S.; Lee, H.K. The autophagy Protein Atg5 Plays a Crucial Role in the Maintenance and Reconstitution Ability of Hematopoietic Stem Cells. Immune Netw. 2019, 19, e12. [Google Scholar] [CrossRef]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Ito, C.; Ooshio, T.; Tadokoro, Y.; Kohno, S.; Ueno, M.; Kobayashi, M.; Kasahara, A.; Takase, Y.; Kurayoshi, K.; et al. Essential role of autophagy in protecting neonatal haematopoietic stem cells from oxidative stress in a p62-independent manner. Sci. Rep. 2021, 11, 1666. [Google Scholar] [CrossRef] [PubMed]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Audesse, A.J.; Dhakal, S.; Hassell, L.-A.; Gardell, Z.; Nemtsova, Y.; Webb, A.E. FOXO3 directly regulates an autophagy network to functionally regulate proteostasis in adult neural stem cells. PLoS Genet. 2019, 15, e1008097. [Google Scholar] [CrossRef]
- Leeman, D.S.; Hebestreit, K.; Ruetz, T.; Webb, A.E.; McKay, A.; Pollina, E.A.; Dulken, B.W.; Zhao, X.; Yeo, R.W.; Ho, T.T.; et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 2018, 359, 1277–1283. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Lapierre, L.R.; Daniel De Magalhaes Filho, C.; McQuary, P.R.; Chu, C.-C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Niederstrasser, H.; Douglas, P.M.; Lin, R.; Jaramillo, J.; Li, Y.; Oswald, N.W.; Zhou, A.; McMillan, E.A.; Mendiratta, S.; et al. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat. Commun. 2017, 8, 2270. [Google Scholar] [CrossRef]
- García-Prat, L.; Kaufmann, K.B.; Schneiter, F.; Voisin, V.; Murison, A.; Chen, J.; Chan-Seng-Yue, M.; Gan, O.I.; McLeod, J.L.; Smith, S.A.; et al. TFEB-mediated endolysosomal activity controls human hematopoietic stem cell fate. Cell Stem Cell 2021, 28, 1838–1850 e10. [Google Scholar] [CrossRef]
- Yuizumi, N.; Harada, Y.; Kuniya, T.; Sunabori, T.; Koike, M.; Wakabayashi, M.; Ishihama, Y.; Suzuki, Y.; Kawaguchi, D.; Gotoh, Y. Maintenance of neural stem-progenitor cells by the lysosomal biosynthesis regulators TFEB and TFE3 in the embryonic mouse telencephalon. Stem Cells 2021, 39, 929–944. [Google Scholar] [CrossRef]
- Kobayashi, T.; Piao, W.; Takamura, T.; Kori, H.; Miyachi, H.; Kitano, S.; Iwamoto, Y.; Yamada, M.; Imayoshi, I.; Shioda, S.; et al. Enhanced lysosomal degradation maintains the quiescent state of neural stem cells. Nat. Commun. 2019, 10, 5446. [Google Scholar] [CrossRef]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef]
- Lin, X.X.; Sen, I.; Janssens, G.E.; Zhou, X.; Fonslow, B.R.; Edgar, D.; Stroustrup, N.; Swoboda, P.; Yates, J.R., 3rd; Ruvkun, G.; et al. DAF-16/FOXO and HLH-30/TFEB function as combinatorial transcription factors to promote stress resistance and longevity. Nat. Commun. 2018, 9, 4400. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Liang, R.; Rimmelé, P.; Bigarella, C.L.; Yalcin, S.; Ghaffari, S. Evidence for AKT-independent regulation of FOXO1 and FOXO3 in haematopoietic stem and progenitor cells. Cell Cycle 2016, 15, 861–867. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, S.; Iwama, A.; Takayanagi, S.-I.; Morita, Y.; Eto, K.; Ema, H.; Nakauchi, H. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 2006, 25, 3515–3523. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a Is Essential for Maintenance of the Hematopoietic Stem Cell Pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, S.; Zhang, X.; Luciano, J.P.; Mungamuisk, S.K.; Marinkovic, D.; Vercherat, C.; Sarkar, A.; Grisotto, M.; Taneja, R.; Ghaffari, S. Foxo3 Is Essential for the Regulation of Ataxia Telangiectasia Mutated and Oxidative Stress-mediated Homeostasis of Hematopoietic Stem Cells. J. Biol. Chem. 2008, 283, 25692–25705. [Google Scholar] [CrossRef] [Green Version]
- Rimmelé, P.; Liang, R.; Bigarella, C.L.; Kocabas, F.; Xie, J.; Serasinghe, M.N.; Chipuk, J.E.; Sadek, H.; Zhang, C.C.; Ghaffari, S. Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO 3. EMBO Rep. 2015, 16, 1164–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindarajah, V.; Lee, J.-M.; Solomon, M.; Goddard, B.; Nayak, R.; Nattamai, K.; Geiger, H.; Salomonis, N.; Cancelas, J.A.; Reynaud, D. FOXO activity adaptation safeguards the hematopoietic stem cell compartment in hyperglycemia. Blood Adv. 2020, 4, 5512–5526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yalcin, S.; Lee, D.-F.; Yeh, T.-Y.J.; Lee, S.-M.; Su, J.; Mungamuisk, S.K.; Rimmelé, P.; Kennedy, M.; Sellers, R.; et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat. Cell Biol. 2011, 13, 1092–1099. [Google Scholar] [CrossRef]
- Liu, P.; Liu, K.; Gu, H.; Wang, W.; Gong, J.; Zhu, Y.; Zhao, Q.; Cao, J.; Han, C.; Gao, F.; et al. High autophagic flux guards ESC identity through coordinating autophagy machinery gene program by FOXO1. Cell Death Differ. 2017, 24, 1672–1680. [Google Scholar] [CrossRef] [Green Version]
- McClelland Descalzo, D.L.; Satoorian, T.S.; Walker, L.M.; Sparks, N.R.; Pulyanina, P.Y.; Zur Nieden, N.I. Glucose-Induced Oxidative Stress Reduces Proliferation in Embryonic Stem Cells via FOXO3A/beta-Catenin-Dependent Transcription of p21(cip1). Stem Cell Rep. 2016, 7, 55–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Prat, L.; Perdiguero, E.; Alonso-Martín, S.; Dell’Orso, S.; Ravichandran, S.; Brooks, S.R.; Juan, A.H.; Campanario, S.; Jiang, K.; Hong, X.; et al. FoxO maintains a genuine muscle stem-cell quiescent state until geriatric age. Nat. Cell Biol. 2020, 22, 1307–1318. [Google Scholar] [CrossRef]
- Gopinath, S.D.; Webb, A.E.; Brunet, A.; Rando, T.A. FOXO3 Promotes Quiescence in Adult Muscle Stem Cells during the Process of Self-Renewal. Stem Cell Rep. 2014, 2, 414–426. [Google Scholar] [CrossRef] [Green Version]
- Signer, R.A.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef]
- Signer, R.A.; Qi, L.; Zhao, Z.; Thompson, D.; Sigova, A.A.; Fan, Z.P.; DeMartino, G.N.; Young, R.A.; Sonenberg, N.; Morrison, S.J. The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E-BPs. Genes Dev. 2016, 30, 1698–1703. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Hoshii, T.; Tadokoro, Y.; Naka, K.; Ooshio, T.; Muraguchi, T.; Sugiyama, N.; Soga, T.; Araki, K.; Yamamura, K.-I.; Hirao, A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Investig. 2012, 122, 2114–2129. [Google Scholar] [CrossRef] [PubMed]
- Hoshii, T.; Matsuda, S.; Hirao, A. Pleiotropic roles of mTOR complexes in haemato-lymphopoiesis and leukemogenesis. J. Biochem. 2014, 156, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, B.; Sahin, E.; Jiang, S.; Sanchez-Aguilera, A.; Scott, K.L.; Chin, L.; Williams, D.A.; Kwiatkowski, D.J.; DePinho, R.A. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc. Natl. Acad. Sci. USA 2008, 105, 19384–19389. [Google Scholar] [CrossRef] [Green Version]
- Campbell, T.B.; Basu, S.; Hangoc, G.; Tao, W.; Broxmeyer, H.E. Overexpression of Rheb2 enhances mouse hematopoietic progenitor cell growth while impairing stem cell repopulation. Blood 2009, 114, 3392–3401. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.; Gilliland, D.G.; Morrison, S.J. mTOR Activation Induces Tumor Suppressors that Inhibit Leukemogenesis and Deplete Hematopoietic Stem Cells after Pten Deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef] [Green Version]
- Kalaitzidis, D.; Sykes, S.M.; Wang, Z.; Punt, N.; Tang, Y.; Ragu, C.; Sinha, A.U.; Lane, S.W.; Souza, A.L.; Clish, C.B.; et al. mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell 2012, 11, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Kasada, A.; Ueno, M.; Hoshii, T.; Tadokoro, Y.; Nomura, N.; Ito, C.; Takase, Y.; Vu, H.T.; Kobayashi, M.; et al. Distinct roles of Rheb and Raptor in activating mTOR complex 1 for the self-renewal of hematopoietic stem cells. Biochem. Biophys. Res. Commun. 2017, 495, 1129–1135. [Google Scholar] [CrossRef]
- Luo, Y.; Li, L.; Zou, P.; Wang, J.; Shao, L.; Zhou, D.; Liu, L. Rapamycin Enhances Long-Term Hematopoietic Reconstitution of Ex Vivo Expanded Mouse Hematopoietic Stem Cells by Inhibiting Senescence. Transplantation 2014, 97, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Rohrabaugh, S.L.; Campbell, T.B.; Hangoc, G.; Broxmeyer, H.E. Ex vivo rapamycin treatment of human cord blood CD34+ cells enhances their engraftment of NSG mice. Blood Cells, Mol. Dis. 2011, 46, 318–320. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Nguyen-McCarty, M.; O Hexner, E.; Danet-Desnoyers, G.; Klein, P.S. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat. Med. 2012, 18, 1778–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2009, 2, ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 2021, 23, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.V.; Tano, M.D.; Mattson, M.P.; Guidi, N. Intermittent and periodic fasting, longevity and disease. Nat. Aging 2021, 1, 47–59. [Google Scholar] [CrossRef]
- Lazare, S.; Ausema, A.; Reijne, A.C.; van Dijk, G.; van Os, R.; de Haan, G. Lifelong dietary intervention does not affect hematopoietic stem cell function. Exp. Hematol. 2017, 53, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Tao, S.; Chen, Z.; Koliesnik, I.O.; Calmes, P.G.; Hoerr, V.; Han, B.; Gebert, N.; Zornig, M.; Loffler, B.; et al. Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J. Exp. Med. 2016, 213, 535–553. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Young, K.; Eudy, E.; Bell, R.; A Loberg, M.; Stearns, T.; Sharma, D.; Velten, L.; Haas, S.; Filippi, M.-D.; Trowbridge, J.J. Decline in IGF1 in the bone marrow microenvironment initiates hematopoietic stem cell aging. Cell Stem Cell 2021, 28, 1473–1482. [Google Scholar] [CrossRef]
- Lee, J.-M.; Govindarajah, V.; Goddard, B.; Hinge, A.; Muench, D.E.; Filippi, M.-D.; Aronow, B.; Cancelas, J.A.; Salomonis, N.; Grimes, H.L.; et al. Obesity alters the long-term fitness of the hematopoietic stem cell compartment through modulation of Gfi1 expression. J. Exp. Med. 2018, 215, 627–644. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef] [Green Version]
- Berg, S.M.V.D.; Seijkens, T.T.P.; Kusters, P.J.H.; Beckers, L.; Toom, M.D.; Smeets, E.; Levels, J.; de Winther, M.P.J.; Lutgens, E. Diet-induced obesity in mice diminishes hematopoietic stem and progenitor cells in the bone marrow. FASEB J. 2016, 30, 1779–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, K.; DelProposto, J.; Morris, D.L.; Zamarron, B.; Mergian, T.; Maley, N.; Cho, K.W.; Geletka, L.; Subbaiah, P.; Muir, L.; et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol. Metab. 2014, 3, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Chen, M.; Kumar, R.; Stefanovic-Racic, M.; O’Doherty, R.M.; Ding, Y.; Jahnen-Dechent, W.; Borghesi, L. Bone marrow lympho-myeloid malfunction in obesity requires precursor cell-autonomous TLR4. Nat. Commun. 2018, 9, 708. [Google Scholar] [CrossRef]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; More, K.J.; Breitbart, R.E.; et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in db/db Mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Masamoto, Y.; Arai, S.; Sato, T.; Kubota, N.; Takamoto, I.; Kadowaki, T.; Kurokawa, M. Adiponectin Enhances Quiescence Exit of Murine Hematopoietic Stem Cells and Hematopoietic Recovery Through mTORC1 Potentiation. Stem Cells 2017, 35, 1835–1848. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.-M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schurmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e6. [Google Scholar] [CrossRef] [Green Version]
- Hermetet, F.; Buffiere, A.; Aznague, A.; Pais de Barros, J.P.; Bastie, J.N.; Delva, L.; Quere, R. High-fat diet disturbs lipid raft/TGF-beta signaling-mediated maintenance of hematopoietic stem cells in mouse bone marrow. Nat. Commun. 2019, 10, 523. [Google Scholar] [CrossRef]
- Yamazaki, S.; Iwama, A.; Takayanagi, S.I.; Eto, K.; Ema, H.; Nakauchi, H. TGF-{beta} as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 2009, 113, 1250–1256. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, S.R. Biophysical and biochemical mechanisms by which dietary N-3 polyunsaturated fatty acids from fish oil disrupt membrane lipid rafts. J. Nutr. Biochem. 2012, 23, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadokoro, Y.; Hoshii, T.; Yamazaki, S.; Eto, K.; Ema, H.; Kobayashi, M.; Ueno, M.; Ohta, K.; Arai, Y.; Hara, E.; et al. Spred1 Safeguards Hematopoietic Homeostasis against Diet-Induced Systemic Stress. Cell Stem Cell 2018, 22, 713–725.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Chen, G.-L.; Hannemann, N.; Ipseiz, N.; Krönke, G.; Bäuerle, T.; Munos, L.; Wirtz, S.; Schett, G.; Bozec, A. Microbiota from Obese Mice Regulate Hematopoietic Stem Cell Differentiation by Altering the Bone Niche. Cell Metab. 2015, 22, 886–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasmant, E.; Gilbert-Dussardier, B.; Petit, A.; De Laval, B.; Luscan, A.; Gruber, A.; Lapillonne, H.; Deswarte, C.; Goussard, P.; Laurendeau, I.; et al. SPRED1, a RAS MAPK pathway inhibitor that causes Legius syndrome, is a tumour suppressor downregulated in paediatric acute myeloblastic leukaemia. Oncogene 2015, 34, 631–638. [Google Scholar] [CrossRef]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef]
- Mana, M.D.; Hussey, A.M.; Tzouanas, C.N.; Imada, S.; Millan, Y.B.; Bahceci, D.; Saiz, D.R.; Webb, A.T.; Lewis, C.A.; Carmeliet, P.; et al. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep. 2021, 35, 109212. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Cheng, C.-W.; Cao, A.; Tripathi, S.; Mana, M.D.; Bauer-Rowe, K.E.; Abu-Remaileh, M.; Clavain, L.; Erdemir, A.; Lewis, C.A.; et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell 2018, 22, 769–778.e4. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.-W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131.e15. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Tu, B.P. Acetyl-CoA and the regulation of metabolism: Mechanisms and consequences. Curr. Opin. Cell. Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, A.; Clément, K.; Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 2021, 70, 1174–1182. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Mainali, R.; Ahmadi, S.; Wang, S.; Singh, R.; Kavanagh, K.; Kitzman, D.W.; Kushugulova, A.; Marotta, F.; Yadav, H. Gut microbiome and aging: Physiological and mechanistic insights. Nutr. Health Aging 2018, 4, 267–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigurdsson, V.; Takei, H.; Soboleva, S.; Radulovic, V.; Galeev, R.; Siva, K.; Leeb-Lundberg, L.M.; Iida, T.; Nittono, H.; Miharada, K. Bile Acids Protect Expanding Hematopoietic Stem Cells from Unfolded Protein Stress in Fetal Liver. Cell Stem Cell 2016, 18, 522–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigurdsson, V.; Haga, Y.; Takei, H.; Mansell, E.; Okamatsu-Haga, C.; Suzuki, M.; Radulovic, V.; Van Der Garde, M.; Koide, S.; Soboleva, S.; et al. Induction of blood-circulating bile acids supports recovery from myelosuppressive chemotherapy. Blood Adv. 2020, 4, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Persaud, A.K.; Nair, S.; Rahman, F.; Raj, R.; Weadick, B.; Nayak, D.; McElroy, C.; Shanmugam, M.; Knoblaugh, S.; Cheng, X.; et al. Facilitative lysosomal transport of bile acids alleviates ER stress in mouse hematopoietic precursors. Nat. Commun. 2021, 12, 1248. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Ryu, S.H.; Koues, O.I.; Collins, P.L.; Solnica-Krezel, L.; Pearce, E.J.; Pearce, E.L.; Oltz, E.M.; Stappenbeck, T.S. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 2016, 167, 1137. [Google Scholar] [CrossRef]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β–FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.; Bruce, J.P.; Wintersinger, J.A.; Mer, A.S.; Lo, E.B.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E.; Simoes, R.D.M.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256.e11. [Google Scholar] [CrossRef]
- Scognamiglio, R.; Cabezas-Wallscheid, N.; Thier, M.; Altamura, S.; Reyes, A.; Prendergast, Á.; Baumgärtner, D.; Carnevalli, L.; Atzberger, A.; Haas, S.; et al. Myc depletion induces a pluripotent dormant state mimicking diapause. Cell 2018, 164, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Bulut-Karslioglu, A.; Biechele, S.; Jin, H.; Macrae, T.A.; Hejna, M.; Gertsenstein, M.; Song, J.S.; Ramalho-Santos, M. Inhibition of mTOR induces a paused pluripotent state. Nature 2016, 540, 119–123. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tadokoro, Y.; Hirao, A. The Role of Nutrients in Maintaining Hematopoietic Stem Cells and Healthy Hematopoiesis for Life. Int. J. Mol. Sci. 2022, 23, 1574. https://doi.org/10.3390/ijms23031574
Tadokoro Y, Hirao A. The Role of Nutrients in Maintaining Hematopoietic Stem Cells and Healthy Hematopoiesis for Life. International Journal of Molecular Sciences. 2022; 23(3):1574. https://doi.org/10.3390/ijms23031574
Chicago/Turabian StyleTadokoro, Yuko, and Atsushi Hirao. 2022. "The Role of Nutrients in Maintaining Hematopoietic Stem Cells and Healthy Hematopoiesis for Life" International Journal of Molecular Sciences 23, no. 3: 1574. https://doi.org/10.3390/ijms23031574