
Apoptosis during ZIKA Virus Infection: Too Soon or Too Late?

 , , , and
, , , and
Abstract
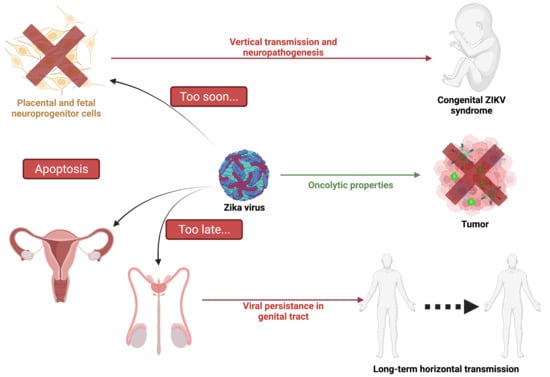
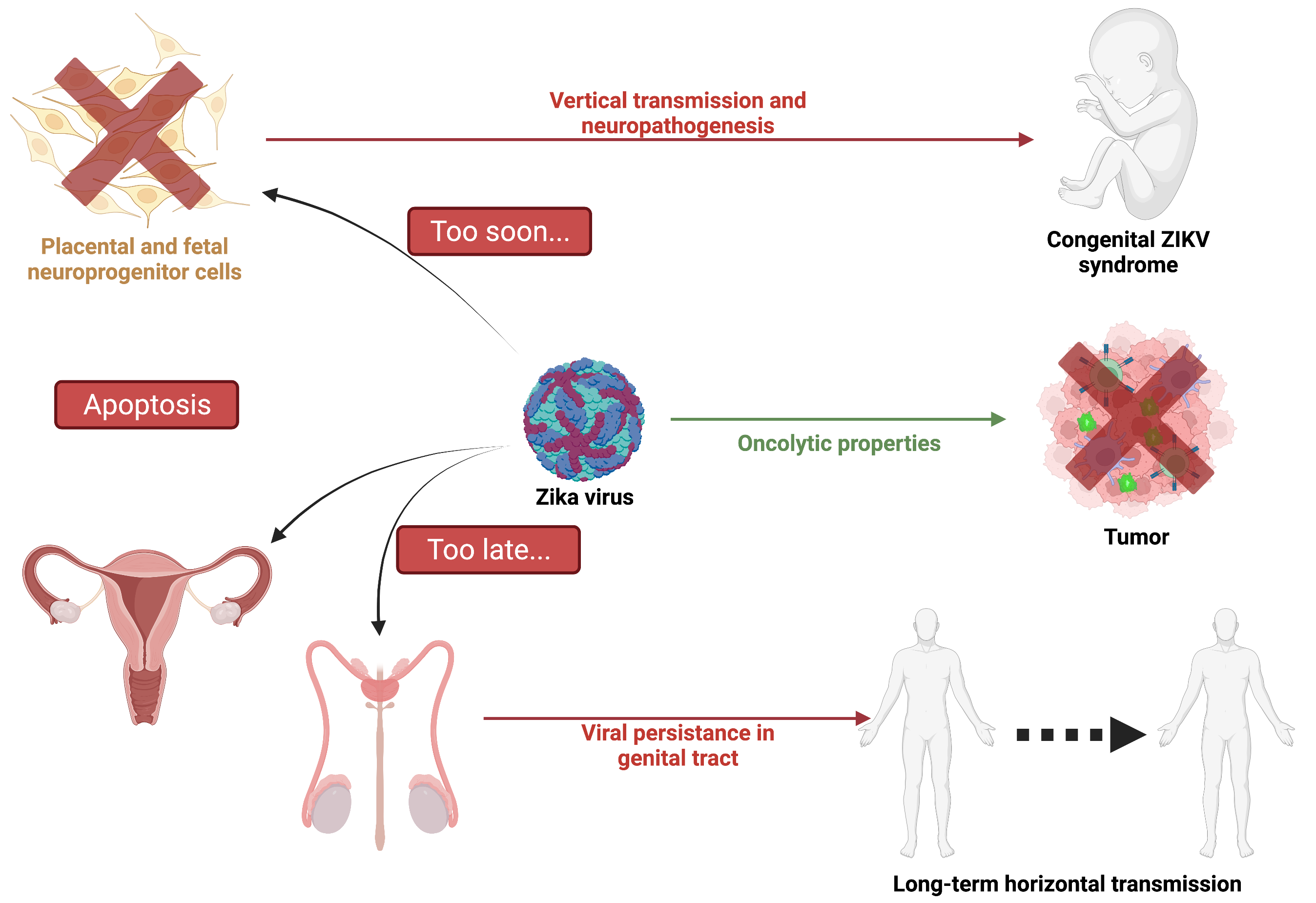
:
1. Introduction
2. Apoptotic Cell Death
2.1. Features of Apoptosis
2.2. Virus-Induced Apoptosis
3. Zika Infection
3.1. Zika Virus, Historical Data
3.2. Zika Virus Transmission and Clinical Outcomes
3.3. Zika Virus Structure and Life Cycle
3.4. Zika Virus Tropism and Pathogenesis
4. Zika Virus and Apoptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Strain | Cellular Model | Apoptotic Cues | Apoptosis and ZIKV Infection Interplay | Reference |
|---|---|---|---|---|
| MR766: East African strain isolated from sentinel rhesus in Uganda in 1947 (Dick, Kitchen, and Haddow 1952) | HT1080 (Human epithelial cells derived from fibrosarcoma) | Apoptotic cell death markers were cleavage of Caspase 3 and PARP. | Apoptosis was shown to be delayed by inhibition of the JAK-STAT pathway by ZIKV NS2B/3 proteins. | [82] |
| Brazil 2015 (KU940228) Brazilian strain 2015 (Calvet et al., 2016) | hNPCs (Human Neural Progenitor cells) | Early apoptosis was induced at 24 h.p.i. with caspase 8, 9, and 3 activation. This viral strain was found to be highly deleterious to human neural progenitor cells. | Apoptosis was shown to limit viral production. This was reversed by the use of Z-VAD which induced an increase in intracellular viral RNA. | [83] |
| MR766 | hNPCs | Apoptotic cell death was induced at 72 h.p.i. Caspase 3 expression was highly increased 3 d.p.i. | N.D. | [84] |
| cDNA encoding the E, prM-E and M-E proteins of the Haitian ZIKV strain (KU509998.3) isolated in 2014 (Lednicky et al., 2016) | PC12 cells (Rat pheochromocytoma cells) | Intrinsic mitochondrial pathway. | Envelope viral protein induces apoptosis by increasing BAX expression and decreasing Bcl-2 expression at the transcriptional and translational levels at 48 h.p.t. | [85] |
| H/PF/2013: French Polynesian 2013 clinical isolate (Cao-Lormeau et al., 2014) | A549 (Human Adenocarcinomic human alveolar basal epithelial cell) | ZIKV induces mitochondrial apoptosis 48 h.p.i. by activating caspase 9 and 3. | Apoptosis is detected when the viral progeny reaches the peak. | [81] |
| MR766 | HuH7 (Human hepatoma cell lines) and BCLXKO HuH7 cells | MCL1 expression decreases in cells infected with ZIKV while BCLXL expression is not affected. BCLXL down-regulation induces cell apoptosis. | Decreased MCL1 expression during ZIKV infection promotes viral replication in vitro. | [86] |
| H/PF/2013 MR766 and Brazil 2015 BeH819015 (molecular clones) | A549, U251MG (derived from a human malignant glioblastoma), HEK293 (Human embryonic kidney 293 cells) | ZIKV infection leads to mitochondrial apoptosis when most of the ZIKV progeny is released by the infected cells. (48 h.p.i), | ZIKV delays apoptosis in infected cells and confers protection against exogenous apoptosis induced by either intrinsic or extrinsic pathways. | [87] |
| PLCal ZV: Asian strain isolated in 2013 in Thailand (Ellison et al., 2016) PRVABC-59: Asian strain isolated in Puerto Rico in 2015 (Lanciotti et al., 2016) | HFAs (Human fetal astrocytes), A549 | Late apoptosis was induced in ZIKV HFA infected cells. Under 50% of the infected HFAs exhibited apoptosis compared to more than 90% for A549 5 d.p.i. This indicates that HFAs are remarkably resistant to apoptosis induced by ZIKV. | Although HFAs have a strong antiviral response, it has been shown that they keep excreting ZIKV for up to 28 d.p.i. | [88] |
| MR766 (molecular clone) PRVABC-59 | Primary human Sertoli cells | Less than 10% of Sertoli PRVABC59 infected cells are apoptotic versus around 70% of A549 infected cells at 72 h.p.i. These percentages are half as low when cells are infected with the MR766 strain. The low level of ZIKV-induced apoptosis detected in Sertoli cells explains the persistence of both American and African ZIKV strains in these cells. | The limited percentage of apoptosis observed in ZIKV-infected Sertoli cells allows the virus to replicate furthermore. The peak of viremia was detected between 3 and 4 d.p.i. | [89] |
| r-MRV (recombinant MR766 strain) PRVABC-59 | HTR-8 cells (Human immortalized first-trimester placental trophoblast cells), JEG-3 and JAR (choriocarcinoma-derived third-trimester placental trophoblast cell lines) | Apoptosis was induced in all three cell lines at 48 h.p.i. CHOP upregulation and nuclear translocation were observed 24 h.p.i. Trophoblast induced-apoptosis involved activation of caspases, ER-Stress markers and most importantly JNK protein. | Apoptosis was strongly inhibited by the use of JNK inhibitors. | [90] |
| MR766, SZ01 (Asian strain, isolated in China in 2016) | hRPTEpiCs human renal proximal tubular epithelial cells | MR766 induced a higher degree of cell apoptosis (48 h.p.i.) compared to SZ01 (9 d.p.i.) | ZIKV persisted for more than 30 d.p.i within the hRPTEpiCs. | [91] |
4.1. Is Early Apoptosis in Development Responsible for the Irreversible Damage Produced by ZIKV Infection?
4.2. Is Delayed and Impaired Apoptosis Responsible for ZIKV Persistence and Unusual Transmission Pathways?
4.3. Apoptosis in the Mosquito Vectors
5. Therapeutics Related to ZIKV, A Role for Apoptosis?
5.1. Antiviral Treatment through Apoptosis Remediation
5.2. ZIKV as Oncolytic Virotherapy
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ZIKV | Zika virus |
| PCD | Programmed cell death |
| CNS | Central Nervous System |
| ER | Endoplasmic Reticulum |
| MOMP | Mitochondrial outer membrane permeabilization |
| APAF-1 | Apoptosis protease activating factor 1 |
| PARP | Poly ADP-ribose polymerase |
| DISC | Death inducing signaling complex |
| UPR | Unresolved unfolded protein response |
| CHOP | C/EBP HOmologous Protein |
| BHV | Bovine herpesvirus 1 |
| DENV | Dengue virus |
| WNV | West Nile virus |
| JEV | Japanese encephalitis virus |
| HSV | Herpes simplex virus HSV |
| MAVS | Mitochondrial antiviral-signaling protein |
| WHO | World Health Organization |
| CDC | Center for disease control |
| CZS | Congenital Zika syndrome |
| sfRNA | Small flavivirus RNA |
| CHIKV | Chikungunya virus |
| RRV | Ross River virus |
References
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.-Y.; Vasilakis, N. Zika Virus: History, Emergence, Biology, and Prospects for Control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martínez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N.; Group, W.Z.C.W. Zika Virus Infection as a Cause of Congenital Brain Abnormalities and Guillain–Barré Syndrome: Systematic Review. PLOS Med. 2017, 14, e1002203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plourde, A.R.; Bloch, E.M. A Literature Review of Zika Virus. Emerg. Infect Dis. 2016, 22, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.J. Viruses and Apoptosis. Int. J. Exp. Pathol. 2001, 82, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.D.; Devitt, A. The Macrophage and the Apoptotic Cell: An Innate Immune Interaction Viewed Simplistically? Immunology 2004, 113, 1–14. [Google Scholar] [CrossRef]
- Thimme, R.; Oldach, D.; Chang, K.-M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of Viral Clearance and Persistence during Acute Hepatitis C Virus Infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef]
- Huang, W.-C.; Abraham, R.; Shim, B.-S.; Choe, H.; Page, D.T. Zika Virus Infection during the Period of Maximal Brain Growth Causes Microcephaly and Corticospinal Neuron Apoptosis in Wild Type Mice. Sci. Rep. 2016, 6, 34793. [Google Scholar] [CrossRef] [Green Version]
- Ximenes, R.; Ramsay, L.C.; Miranda, R.N.; Morris, S.K.; Murphy, K.; Sander, B. Health Outcomes Associated with Zika Virus Infection in Humans: A Systematic Review of Systematic Reviews. BMJ Open 2019, 9, e032275. [Google Scholar] [CrossRef] [Green Version]
- Stassen, L.; Armitage, C.; van der Heide, D.; Beagley, K.; Frentiu, F. Zika Virus in the Male Reproductive Tract. Viruses 2018, 10, 198. [Google Scholar] [CrossRef] [Green Version]
- Meier, P.; Finch, A.; Evan, G. Apoptosis in Development. Nature 2000, 407, 796–801. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, New and Emerging Functions of Caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, T.; Martinou, J.-C. Where Killers Meet--Permeabilization of the Outer Mitochondrial Membrane during Apoptosis. Cold Spring Harb. Perspect. Biol. 2013, 5, a011106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Li, Y.; Hu, Q.; Bai, X.; Huang, W.; Yan, C.; Scheres, S.H.W.; Shi, Y. Atomic Structure of the Apoptosome: Mechanism of Cytochrome c—And DATP-Mediated Activation of Apaf-1. Genes Dev. 2015, 29, 2349–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malladi, S.; Challa-Malladi, M.; Fearnhead, H.O.; Bratton, S.B. The Apaf-1•procaspase-9 Apoptosome Complex Functions as a Proteolytic-Based Molecular Timer. EMBO J. 2009, 28, 1916–1925. [Google Scholar] [CrossRef] [Green Version]
- Tummers, B.; Green, D.R. Caspase-8: Regulating Life and Death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Stennicke, H.R.; Jürgensmeier, J.M.; Shin, H.; Deveraux, Q.; Wolf, B.B.; Yang, X.; Zhou, Q.; Ellerby, H.M.; Ellerby, L.M.; Bredesen, D.; et al. Pro-Caspase-3 Is a Major Physiologic Target of Caspase-8. J. Biol. Chem. 1998, 273, 27084–27090. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER Stress-Induced Cell Death Mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhang, K.; Huang, Y.; Ding, L.; Chen, G.; Zhang, H.; Tong, D. Bovine Herpes Virus Type 1 Induces Apoptosis through Fas-Dependent and Mitochondria-Controlled Manner in Madin-Darby Bovine Kidney Cells. Virol. J. 2012, 9, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Bacha, T.; Midlej, V.; Pereira da Silva, A.P.; Silva da Costa, L.; Benchimol, M.; Galina, A.; Da Poian, A.T. Mitochondrial and Bioenergetic Dysfunction in Human Hepatic Cells Infected with Dengue 2 Virus. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2007, 1772, 1158–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, T.X.; Randall, G. Flavivirus Modulation of Cellular Metabolism. Curr. Opin. Virol. 2016, 19, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Thaker, S.K.; Ch’ng, J.; Christofk, H.R. Viral Hijacking of Cellular Metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef]
- Wang, Q.; Xin, X.; Wang, T.; Wan, J.; Ou, Y.; Yang, Z.; Yu, Q.; Zhu, L.; Guo, Y.; Wu, Y.; et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J. Virol. 2019, 93, e00887-19. [Google Scholar] [CrossRef] [Green Version]
- Johnston, B.P.; McCormick, C. Herpesviruses and the Unfolded Protein Response. Viruses 2019, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Singh, N.; Sengupta, N.; Fatima, M.; Seth, P.; Mahadevan, A.; Shankar, S.K.; Bhattacharyya, A.; Basu, A. Japanese Encephalitis Virus Induces Human Neural Stem/Progenitor Cell Death by Elevating GRP78, PHB and HnRNPC through ER Stress. Cell Death Dis. 2018, 8, e2556. [Google Scholar] [CrossRef] [Green Version]
- Catteau, A.; Roué, G.; Yuste, V.J.; Susin, S.A.; Desprès, P. Expression of Dengue ApoptoM Sequence Results in Disruption of Mitochondrial Potential and Caspase Activation. Biochimie 2003, 85, 789–793. [Google Scholar] [CrossRef]
- Vanwalscappel, B.; Haddad, J.G.; Almokdad, R.; Decotter, J.; Gadea, G.; Desprès, P. Zika M Oligopeptide ZAMP Confers Cell Death-Promoting Capability to a Soluble Tumor-Associated Antigen through Caspase-3/7 Activation. Int. J. Mol. Sci. 2020, 21, 9578. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Cheng, A.; Wang, M.; Yin, Z.; Jia, R. The Dual Regulation of Apoptosis by Flavivirus. Front. Microbiol. 2021, 12, 654494. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Suzuki, T.; Kusakabe, S.; Tokunaga, M.; Hirano, J.; Miyata, Y.; Matsuura, Y. Regulation of Apoptosis during Flavivirus Infection. Viruses 2017, 9, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, X.; Li, Y.; Qi, Y.; Xu, J.; Wang, Z.; Zhang, X.; Zhang, D.; Zhang, L.; Huang, J. XAF1 Contributes to Dengue Virus-Induced Apoptosis in Vascular Endothelial Cells. FASEB J. 2013, 27, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen Parquet, M.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile Virus-Induced Bax-Dependent Apoptosis. FEBS Lett. 2001, 500, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Maelfait, J.; Liverpool, L.; Rehwinkel, J. Nucleic Acid Sensors and Programmed Cell Death. J. Mol. Biol. 2020, 432, 552–568. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Chiang, R.-L.; Chang, T.-H.; Liao, C.-L.; Lin, Y.-L. The Interferon Stimulator Mitochondrial Antiviral Signaling Protein Facilitates Cell Death by Disrupting the Mitochondrial Membrane Potential and by Activating Caspases. J. Virol. 2010. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Tschopp, J. IRF-3 Partners Bax in a Viral-Induced Dance Macabre. EMBO J. 2010, 29, 1627–1628. [Google Scholar] [CrossRef] [Green Version]
- Knowlton, J.J.; Dermody, T.S.; Holm, G.H. Apoptosis Induced by Mammalian Reovirus Is Beta Interferon (IFN) Independent and Enhanced by IFN Regulatory Factor 3- and NF-ΚB-Dependent Expression of Noxa. J. Virol. 2012, 86, 1650–1660. [Google Scholar] [CrossRef] [Green Version]
- Valadão, A.L.C.; Aguiar, R.S.; de Arruda, L.B. Interplay between Inflammation and Cellular Stress Triggered by Flaviviridae Viruses. Front. Microbiol. 2016, 7, 1233. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.N.; Kanneganti, T.-D. PANoptosis in Viral Infection: The Missing Puzzle Piece in the Cell Death Field. J. Mol. Biol. 2021, 167249. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.; Calvez, E.; Chouin-Carneiro, T.; Diallo, D.; Failloux, A.-B. An Overview of Mosquito Vectors of Zika Virus. Microbes Infect. 2018, 20, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W.A.; Kitchen, S.F.; Haddow, A.J. Zika Virus (I). Isolations and Serological Specificity. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Smithburn, K.C. Neutralizing Antibodies Against Certain Recently Isolated Viruses in the Sera of Human Beings Residing in East Africa. J. Immunol. 1952, 69, 223–234. [Google Scholar] [PubMed]
- Duffy, M.R.; Chen, T.-H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika Virus Outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.-M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.-L.; Mallet, H.-P.; Sall, A.A.; Musso, D. Zika Virus, French Polynesia, South Pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1084–1086. [Google Scholar] [CrossRef]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika Virus Outbreak, Bahia, Brazil. Emerg. Infect. Dis. 2015, 21, 1885–1886. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, Z.-Y.; Han, J.-F.; Jiang, T.; Li, X.-F.; Qin, C.-F. Genomic Characterization and Phylogenetic Analysis of Zika Virus Circulating in the Americas. Infect. Genet. Evol. 2016, 43, 43–49. [Google Scholar] [CrossRef]
- Yadav, P.D.; Malhotra, B.; Sapkal, G.; Nyayanit, D.A.; Deshpande, G.; Gupta, N.; Padinjaremattathil, U.T.; Sharma, H.; Sahay, R.R.; Sharma, P.; et al. Zika Virus Outbreak in Rajasthan, India in 2018 Was Caused by a Virus Endemic to Asia. Infect. Genet. Evol. 2019, 69, 199–202. [Google Scholar] [CrossRef]
- Giron, S.; Franke, F.; Decoppet, A.; Cadiou, B.; Travaglini, T.; Thirion, L.; Durand, G.; Jeannin, C.; L’Ambert, G.; Grard, G.; et al. Vector-Borne Transmission of Zika Virus in Europe, Southern France, August 2019. Eurosurveillance 2019, 24, 1900655. [Google Scholar] [CrossRef] [Green Version]
- Information for Travellers Visiting Countries with Zika Virus Transmission. Available online: https://www.who.int/publications/m/item/information-for-travellers-visiting-countries-with-zika-virus-transmission (accessed on 21 December 2021).
- McKenzie, B.A.; Wilson, A.E.; Zohdy, S. Aedes Albopictus Is a Competent Vector of Zika Virus: A Meta-Analysis. PLoS ONE 2019, 14, e0216794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counotte, M.J.; Kim, C.R.; Wang, J.; Bernstein, K.; Deal, C.D.; Broutet, N.J.N.; Low, N. Sexual Transmission of Zika Virus and Other Flaviviruses: A Living Systematic Review. PLoS Med. 2018, 15, e1002611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO|Sexual Transmission of Zika Virus: Current Status, Challenges and Research Priorities. Available online: http://www.who.int/reproductivehealth/zika/sexual-transmission-experts-meeting/en/ (accessed on 21 December 2021).
- Chan, J.F.W.; Choi, G.K.Y.; Yip, C.C.Y.; Cheng, V.C.C.; Yuen, K.-Y. Zika Fever and Congenital Zika Syndrome: An Unexpected Emerging Arboviral Disease. J. Infect. 2016, 72, 507–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noorbakhsh, F.; Abdolmohammadi, K.; Fatahi, Y.; Dalili, H.; Rasoolinejad, M.; Rezaei, F.; Salehi-Vaziri, M.; Zahra SHAFIEI-JANDAGHI, N.; Shamsi GOOSHKI, E.; Zaim, M.; et al. Zika Virus Infection, Basic and Clinical Aspects: A Review Article. Iran. J. Public Health. 2019. [Google Scholar] [CrossRef] [Green Version]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome Outbreak Associated with Zika Virus Infection in French Polynesia: A Case-Control Study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Correa, J.; de Siqueira, I.C.; Mota, S.; do Rosário, M.S.; Pereira de Jesus, P.A.; Alcantara, L.C.J.; Ernst, J.D.; Rodriguez, A. Anti-Ganglioside Antibodies in Patients with Zika Virus Infection-Associated Guillain-Barré Syndrome in Brazil. PLoS Negl. Trop. Dis. 2019, 13, e0007695. [Google Scholar] [CrossRef] [Green Version]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Da Cunha, A.J.L.A.; de Magalhães-Barbosa, M.C.; Lima-Setta, F.; Medronho, R.; de Andrade, R.; Prata-Barbosae, A. Microcephaly Case Fatality Rate Associated with Zika Virus Infection in Brazil: Current Estimates. Pediatric Infect. Dis. J. 2017, 36, 528–530. [Google Scholar] [CrossRef]
- Costa, M.C.N.; Cardim, L.L.; Teixeira, M.G.; Barreto, M.L.; Carvalho-Sauer, R.d.C.O.; Barreto, F.R.; Carvalho, M.S.I.; Oliveira, W.K.; França, G.V.A.; Carmo, E.H.; et al. Case Fatality Rate Related to Microcephaly Congenital Zika Syndrome and Associated Factors: A Nationwide Retrospective Study in Brazil. Viruses 2020, 12, 1228. [Google Scholar] [CrossRef]
- Paz-Bailey, G.; Rosenberg, E.S.; Doyle, K.; Munoz-Jordan, J.; Santiago, G.A.; Klein, L.; Perez-Padilla, J.; Medina, F.A.; Waterman, S.H.; Adams, L.E.; et al. Persistence of Zika Virus in Body Fluids—Final Report. N. Engl. J. Med. 2018, 379, 1234–1243. [Google Scholar] [CrossRef]
- Campos, G.S.; Hughes Carvalho, R.; Bandeira, A.C.; Reboredo-Oliveira, L.; dos Santos Costa, R.; Figueiredo, C.A.; Sardi, S.I. New Challenge for Zika Virus Infection: Human Reservoirs? Viral Immunol. 2020, 33, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.; Bakkour, S.; Lanteri, M.C.; Brambilla, D.; Simmons, G.; Bruhn, R.; Kaidarova, Z.; Lee, T.-H.; Orlando Alsina, J.; Williamson, P.C.; et al. Zika Virus RNA and IgM Persistence in Blood Compartments and Body Fluids: A Prospective Observational Study. Lancet Infect. Dis. 2020, 20, 1446–1456. [Google Scholar] [CrossRef]
- Petersen, L.R.; Jamieson, D.J.; Powers, A.M.; Honein, M.A. Zika Virus. N. Engl. J. Med. 2016, 374, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, G.; Lagrave, A.; Ogire, E.; Grondin, L.; Seriacaroupin, S.; Moutoussamy, C.; Mavingui, P.; Hoarau, J.-J.; Roche, M.; Krejbich-Trotot, P.; et al. Viral Toxin NS1 Implication in Dengue Pathogenesis Making It a Pivotal Target in Development of Efficient Vaccine. Vaccines 2021, 9, 946. [Google Scholar] [CrossRef]
- Mazeaud, C.; Freppel, W.; Chatel-Chaix, L. The Multiples Fates of the Flavivirus RNA Genome During Pathogenesis. Front. Genet. 2018, 9, 595. [Google Scholar] [CrossRef]
- Lowe, R.; Barcellos, C.; Brasil, P.; Cruz, O.; Honório, N.; Kuper, H.; Carvalho, M. The Zika Virus Epidemic in Brazil: From Discovery to Future Implications. IJERPH 2018, 15, 96. [Google Scholar] [CrossRef] [Green Version]
- de Aragao, M.F.V.; van der Linden, V.; Brainer-Lima, A.M.; Coeli, R.R.; Rocha, M.A.; da Silva, P.S.; de Carvalho, M.D.C.G.; van derLinden, A.; de Holanda, A.C.; Valenca, M.M. Clinical Features and Neuroimaging (CT and MRI) Findings in Presumed Zika Virus Related Congenital Infection and Microcephaly: Retrospective Case Series Study. BMJ 2016, 353, i1901. [Google Scholar] [CrossRef] [Green Version]
- Coyne, C.B.; Lazear, H.M. Zika Virus—Reigniting the TORCH. Nat. Rev. Microbiol. 2016, 14, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Gadea, G.; Bos, S.; Krejbich-Trotot, P.; Clain, E.; Viranaicken, W.; El-Kalamouni, C.; Mavingui, P.; Desprès, P. A Robust Method for the Rapid Generation of Recombinant Zika Virus Expressing the GFP Reporter Gene. Virology 2016, 497, 157–162. [Google Scholar] [CrossRef]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Pena, L.J.; Miranda Guarines, K.; Duarte Silva, A.J.; Sales Leal, L.R.; Mendes Félix, D.; Silva, A.; de Oliveira, S.A.; Junqueira Ayres, C.F.; Júnior, A.S.; de Freitas, A.C. In Vitro and in Vivo Models for Studying Zika Virus Biology. J. Gen. Virol. 2018, 99, 1529–1550. [Google Scholar] [CrossRef] [PubMed]
- Bayless, N.L.; Greenberg, R.S.; Swigut, T.; Wysocka, J.; Blish, C.A. Zika Virus Infection Induces Cranial Neural Crest Cells to Produce Cytokines at Levels Detrimental for Neurogenesis. Cell Host Microbe 2016, 20, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuna, C.E.; Lim, S.-Y.; Deleage, C.; Griffin, B.D.; Stein, D.; Schroeder, L.T.; Omange, R.; Best, K.; Luo, M.; Hraber, P.T.; et al. Zika Viral Dynamics and Shedding in Rhesus and Cynomolgus Macaques. Nat. Med. 2016, 22, 1448–1455. [Google Scholar] [CrossRef] [Green Version]
- Marrazzo, P.; Cricca, M.; Nastasi, C. Are the Organoid Models an Invaluable Contribution to ZIKA Virus Research? Pathogens 2021, 10, 1233. [Google Scholar] [CrossRef]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika Virus Impairs Growth in Human Neurospheres and Brain Organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [Green Version]
- Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Michlmayr, D.; Wang, C.; Fang-Hoover, J.; Harris, E.; Pereira, L. Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell Host Microbe 2016, 20, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Butsabong, T.; Felippe, M.; Campagnolo, P.; Maringer, K. The Emerging Role of Perivascular Cells (Pericytes) in Viral Pathogenesis. J. Gen. Virol. 2021, 102. [Google Scholar] [CrossRef]
- Frumence, E.; Roche, M.; Krejbich-Trotot, P.; El-Kalamouni, C.; Nativel, B.; Rondeau, P.; Missé, D.; Gadea, G.; Viranaicken, W.; Desprès, P. The South Pacific Epidemic Strain of Zika Virus Replicates Efficiently in Human Epithelial A549 Cells Leading to IFN-β Production and Apoptosis Induction. Virology 2016, 493, 217–226. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika Virus Evades Interferon-Mediated Antiviral Response through the Co-Operation of Multiple Nonstructural Proteins in Vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef]
- Souza, B.S.F.; Sampaio, G.L.A.; Pereira, C.S.; Campos, G.S.; Sardi, S.I.; Freitas, L.A.R.; Figueira, C.P.; Paredes, B.D.; Nonaka, C.K.V.; Azevedo, C.M.; et al. Zika Virus Infection Induces Mitosis Abnormalities and Apoptotic Cell Death of Human Neural Progenitor Cells. Sci. Rep. 2016, 6, 39775. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, Q.; Li, X.; Qiu, Z.; Li, A.; Liang, W.; Chen, H.; Cai, X.; Chen, X.; Duan, X.; et al. Zika Virus Envelope Protein Induces G2/M Cell Cycle Arrest and Apoptosis via an Intrinsic Cell Death Signaling Pathway in Neuroendocrine PC12 Cells. Int. J. Biol. Sci. 2018, 14, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Okamoto, T.; Katoh, H.; Sugiyama, Y.; Kusakabe, S.; Tokunaga, M.; Hirano, J.; Miyata, Y.; Fukuhara, T.; Ikawa, M.; et al. Infection with Flaviviruses Requires BCLXL for Cell Survival. PLoS Pathog. 2018, 14, e1007299. [Google Scholar] [CrossRef] [PubMed]
- Turpin, J.; Frumence, E.; Desprès, P.; Viranaicken, W.; Krejbich-Trotot, P. The ZIKA Virus Delays Cell Death Through the Anti-Apoptotic Bcl-2 Family Proteins. Cells 2019, 8, 1338. [Google Scholar] [CrossRef] [Green Version]
- Limonta, D.; Jovel, J.; Kumar, A.; Airo, A.; Hou, S.; Saito, L.; Branton, W.; Ka-Shu Wong, G.; Mason, A.; Power, C.; et al. Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence. Viruses 2018, 10, 646. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Jovel, J.; Lopez-Orozco, J.; Limonta, D.; Airo, A.M.; Hou, S.; Stryapunina, I.; Fibke, C.; Moore, R.B.; Hobman, T.C. Human Sertoli Cells Support High Levels of Zika Virus Replication and Persistence. Sci. Rep. 2018, 8, 5477. [Google Scholar] [CrossRef]
- Muthuraj, P.G.; Sahoo, P.K.; Kraus, M.; Bruett, T.; Annamalai, A.S.; Pattnaik, A.; Pattnaik, A.K.; Byrareddy, S.N.; Natarajan, S.K. Zika Virus Infection Induces Endoplasmic Reticulum Stress and Apoptosis in Placental Trophoblasts. Cell Death Discov. 2021, 7, 24. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.; Chen, J.; Zhou, X.; Dong, Z.; Chen, T.; Yang, Y.; Zou, P.; Jiang, B.; Hu, Y.; et al. Zika Virus Infects Renal Proximal Tubular Epithelial Cells with Prolonged Persistency and Cytopathic Effects. Emerg. Microbes Infect. 2017, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Turpin, J.; El-Safadi, D.; Lebeau, G.; Frumence, E.; Desprès, P.; Viranaïcken, W.; Krejbich-Trotot, P. CHOP Pro-Apoptotic Transcriptional Program in Response to ER Stress Is Hacked by Zika Virus. Int. J. Mol. Sci. 2021, 22, 3750. [Google Scholar] [CrossRef]
- Limonta, D.; Jovel, J.; Kumar, A.; Lu, J.; Hou, S.; Airo, A.M.; Lopez-Orozco, J.; Wong, C.P.; Saito, L.; Branton, W.; et al. Fibroblast Growth Factor 2 Enhances Zika Virus Infection in Human Fetal Brain. J. Infect. Dis. 2019, 220, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liao, H.-M.; Li, B.; Tsai, S.; Hung, G.-C.; Lo, S.-C. Comparative Genomics, Infectivity and Cytopathogenicity of American Isolates of Zika Virus That Developed Persistent Infections in Human Embryonic Kidney (HEK293) Cells. Int. J. Mol. Sci. 2019, 20, 3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, T.; Rajah, M.; Sagan, S. Contemporary Zika Virus Isolates Induce More DsRNA and Produce More Negative-Strand Intermediate in Human Astrocytoma Cells. Viruses 2018, 10, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Wang, J.; Yang, Y.; Qu, S.; Wan, F.; Zhang, Z.; Wang, R.; Li, G.; Cong, H. Zika Virus Infection Induced Apoptosis by Modulating the Recruitment and Activation of Proapoptotic Protein Bax. J. Virol. 2021, 95, e01445-20. [Google Scholar] [CrossRef] [PubMed]
- Auriti, C.; De Rose, D.U.; Santisi, A.; Martini, L.; Piersigilli, F.; Bersani, I.; Ronchetti, M.P.; Caforio, L. Pregnancy and Viral Infections: Mechanisms of Fetal Damage, Diagnosis and Prevention of Neonatal Adverse Outcomes from Cytomegalovirus to SARS-CoV-2 and Zika Virus. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166198. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-F.; Chu, L.-W.; Liao, I.-C.; Simanjuntak, Y.; Lin, Y.-L.; Juan, C.-C.; Ping, Y.-H. The Mechanism of the Zika Virus Crossing the Placental Barrier and the Blood-Brain Barrier. Front. Microbiol. 2020, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L. Congenital Viral Infection: Traversing the Uterine-Placental Interface. Annu. Rev. Virol. 2018, 5, 273–299. [Google Scholar] [CrossRef] [Green Version]
- Rabelo, K.; de Souza, L.J.; Salomão, N.G.; Machado, L.N.; Pereira, P.G.; Portari, E.A.; Basílio-de-Oliveira, R.; Dos Santos, F.B.; Neves, L.D.; Morgade, L.F.; et al. Zika Induces Human Placental Damage and Inflammation. Front. Immunol 2020, 11, 2146. [Google Scholar] [CrossRef]
- Sen Santara, S.; Crespo, Â.C.; Mulik, S.; Ovies, C.; Boulenouar, S.; Strominger, J.L.; Lieberman, J. Decidual NK Cells Kill Zika Virus-Infected Trophoblasts. Proc. Natl. Acad. Sci. USA 2021, 118, e2115410118. [Google Scholar] [CrossRef]
- Ropidi, M.I.M.; Khazali, A.S.; Rashid, N.N.; Yusof, R. Endoplasmic reticulum: A focal point of Zika virus infec-tion. J. Biomed. Sci. 2020, 27, 27. [Google Scholar] [CrossRef] [Green Version]
- Alfano, C.; Gladwyn-Ng, I.; Couderc, T.; Lecuit, M.; Nguyen, L. The Unfolded Protein Response: A Key Player in Zika Virus-Associated Congenital Microcephaly. Front. Cell. Neurosci. 2019, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Gladwyn-Ng, I.; Cordón-Barris, L.; Alfano, C.; Creppe, C.; Couderc, T.; Morelli, G.; Thelen, N.; America, M.; Bessières, B.; Encha-Razavi, F.; et al. Stress-Induced Unfolded Protein Response Contributes to Zika Virus–Associated Microcephaly. Nat. Neurosci. 2018, 21, 63–71. [Google Scholar] [CrossRef]
- Tan, Z.; Zhang, W.; Sun, J.; Fu, Z.; Ke, X.; Zheng, C.; Zhang, Y.; Li, P.; Liu, Y.; Hu, Q.; et al. ZIKV Infection Activates the IRE1-XBP1 and ATF6 Pathways of Unfolded Protein Response in Neural Cells. J. Neuroinflamm. 2018, 15, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, J.; Frumence, E.; Harrabi, W.; Haddad, J.G.; El Kalamouni, C.; Desprès, P.; Krejbich-Trotot, P.; Viranaïcken, W. Zika Virus Subversion of Chaperone GRP78/BiP Expression in A549 Cells during UPR Activation. Biochimie 2020, 175, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER Stress Sensor and Cell Fate Executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP Is Implicated in Programmed Cell Death in Response to Impaired Function of the Endoplasmic Reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Quicke, K.M.; Bowen, J.R.; Johnson, E.L.; McDonald, C.E.; Ma, H.; O’Neal, J.T.; Rajakumar, A.; Wrammert, J.; Rimawi, B.H.; Pulendran, B.; et al. Zika Virus Infects Human Placental Macrophages. Cell Host Microbe 2016, 20, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Michlmayr, D.; Harris, E.; Pereira, L. Zika Virus Replicates in Proliferating Cells in Explants From First-Trimester Human Placentas, Potential Sites for Dissemination of Infection. J. Infect. Dis. 2018, 217, 1202–1213. [Google Scholar] [CrossRef] [Green Version]
- Ghouzzi, V.E.; Bianchi, F.T.; Molineris, I.; Mounce, B.C.; Berto, G.E.; Rak, M.; Lebon, S.; Aubry, L.; Tocco, C.; Gai, M.; et al. ZIKA Virus Elicits P53 Activation and Genotoxic Stress in Human Neural Progenitors Similar to Mutations Involved in Severe Forms of Genetic Microcephaly and P53. Cell Death Dis. 2016, 7, e2440. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.-Y.; Wang, Y.-L.; Wu, W.-L.; Wolseley, V.; Tsai, M.-T.; Radic, V.; Thornton, M.E.; Grubbs, B.H.; Chow, R.H.; Huang, I.-C. Zika Virus Infects Intermediate Progenitor Cells and Post-Mitotic Committed Neurons in Human Fetal Brain Tissues. Sci. Rep. 2017, 7, 14883. [Google Scholar] [CrossRef] [Green Version]
- Schultz, V.; Barrie, J.A.; Donald, C.L.; Crawford, C.L.; Mullin, M.; Anderson, T.J.; Solomon, T.; Barnett, S.C.; Linington, C.; Kohl, A.; et al. Oligodendrocytes Are Susceptible to Zika Virus Infection in a Mouse Model of Perinatal Exposure: Implications for CNS Complications. Glia 2021, 69, 2023–2036. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, X.; Wang, G.; Cheng, X.; Yan, Y.; Fu, Y.; Yang, X.; Jiang, Z. Zika Virus Induces Abnormal Cranial Osteogenesis by Negatively Affecting Cranial Neural Crest Development. Infect. Genet. Evol. 2019, 69, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Martinot, A.J.; Abbink, P.; Afacan, O.; Prohl, A.K.; Bronson, R.; Hecht, J.L.; Borducchi, E.N.; Larocca, R.A.; Peterson, R.L.; Rinaldi, W.; et al. Fetal Neuropathology in Zika Virus-Infected Pregnant Female Rhesus Monkeys. Cell 2018, 173, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mendonça-Vieira, L.R.; Aníbal-Silva, C.E.; Menezes-Neto, A.; de Azevedo, E.A.N.; Zanluqui, N.G.; Peron, J.P.S.; de Oliveira Franca, R.F. Reactive Oxygen Species (ROS) Are Not a Key Determinant for Zika Virus-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells. Viruses 2021, 13, 2111. [Google Scholar] [CrossRef]
- Zhang, F.; Hammack, C.; Ogden, S.C.; Cheng, Y.; Lee, E.M.; Wen, Z.; Qian, X.; Nguyen, H.N.; Li, Y.; Yao, B.; et al. Molecular Signatures Associated with ZIKV Exposure in Human Cortical Neural Progenitors. Nucleic Acids Res. 2016, 44, 8610–8620. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, J.-A.; Oh, S.-J.; Lee, E.-W.; Shin, O.S. Zika Virus Induces Tumor Necrosis Factor-Related Apoptosis Inducing Ligand (TRAIL)-Mediated Apoptosis in Human Neural Progenitor Cells. Cells 2020, 9, E2487. [Google Scholar] [CrossRef]
- Rothan, H.A.; Fang, S.; Mahesh, M.; Byrareddy, S.N. Zika Virus and the Metabolism of Neuronal Cells. Mol. Neurobiol. 2019, 56, 2551–2557. [Google Scholar] [CrossRef]
- Devhare, P.; Meyer, K.; Steele, R.; Ray, R.B.; Ray, R. Zika Virus Infection Dysregulates Human Neural Stem Cell Growth and Inhibits Differentiation into Neuroprogenitor Cells. Cell Death Dis. 2017, 8, e3106. [Google Scholar] [CrossRef] [Green Version]
- Bernhauerová, V.; Rezelj, V.V.; Vignuzzi, M. Modelling Degradation and Replication Kinetics of the Zika Virus In Vitro Infection. Viruses 2020, 12, 547. [Google Scholar] [CrossRef]
- Krejbich-Trotot, P.; Denizot, M.; Hoarau, J.-J.; Jaffar-Bandjee, M.-C.; Das, T.; Gasque, P. Chikungunya Virus Mobilizes the Apoptotic Machinery to Invade Host Cell Defenses. FASEB J. 2011, 25, 314–325. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, J.R.; Azevedo, R.S.S.; Martins Filho, A.J.; Araujo, M.T.F.; Moutinho, E.R.C.; Baldez Vasconcelos, B.C.; Cruz, A.C.R.; Oliveira, C.S.; Martins, L.C.; Baldez Vasconcelos, B.H.; et al. Correlation between Apoptosis and in Situ Immune Response in Fatal Cases of Microcephaly Caused by Zika Virus. Am. J. Pathol. 2018, 188, 2644–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlera, L.; Melik, W.; Bloom, M.E. The Role of Viral Persistence in Flavivirus Biology. Pathog. Dis. 2014, 71, 137–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, C.A.; Norris, P.S.; Ware, C.F. To Kill or Be Killed: Viral Evasion of Apoptosis. Nat. Immunol. 2002, 3, 1013–1018. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Quarleri, J.; Cevallos, C.; Delpino, M.V. Chapter One—Apoptosis in Infectious Diseases as a Mechanism of Immune Evasion and Survival. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Apoptosis in Health and Disease—Part A; Academic Press: Cambridge, MA, USA, 2021; Volume 125, pp. 1–24. [Google Scholar]
- Foy, B.D.; Kobylinski, K.C.; Foy, J.L.C.; Blitvich, B.J.; da Rosa, A.T.; Haddow, A.D.; Lanciotti, R.S.; Tesh, R.B. Probable Non–Vector-Borne Transmission of Zika Virus, Colorado, USA. Emerg. Infect. Dis. 2011, 17, 5. [Google Scholar] [CrossRef]
- Nicastri, E.; Castilletti, C.; Liuzzi, G.; Iannetta, M.; Capobianchi, M.R.; Ippolito, G. Persistent Detection of Zika Virus RNA in Semen for Six Months after Symptom Onset in a Traveller Returning from Haiti to Italy, February 2016. Eurosurveillance 2016, 21, 30314. [Google Scholar] [CrossRef] [Green Version]
- Alcendor, D.J. Zika Virus Infection of the Human Glomerular Cells: Implications for Viral Reservoirs and Renal Pathogenesis. J. Infect. Dis. 2017, 216, 162–171. [Google Scholar] [CrossRef]
- Van der Linden, V.; Pessoa, A.; Dobyns, W.; Barkovich, A.J.; van der Linden, H.L., Jr.; Filho, E.L.R.; Ribeiro, E.M.; Leal, M.; de Araújo, C.; de Maria, F.V.V.; et al. Description of 13 Infants Born During October 2015–January 2016 With Congenital Zika Virus Infection Without Microcephaly at Birth—Brazil. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1343–1348. [Google Scholar] [CrossRef]
- Mulkey, S.B.; Arroyave-Wessel, M.; Peyton, C.; Bulas, D.I.; Fourzali, Y.; Jiang, J.; Russo, S.; McCarter, R.; Msall, M.E.; du Plessis, A.J.; et al. Neurodevelopmental Abnormalities in Children With In Utero Zika Virus Exposure Without Congenital Zika Syndrome. JAMA Pediatr. 2020, 174, 269. [Google Scholar] [CrossRef]
- Palatini, U.; Miesen, P.; Carballar-Lejarazu, R.; Ometto, L.; Rizzo, E.; Tu, Z.; van Rij, R.P.; Bonizzoni, M. Comparative Genomics Shows That Viral Integrations Are Abundant and Express PiRNAs in the Arboviral Vectors Aedes Aegypti and Aedes Albopictus. BMC Genom. 2017, 18, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. Cytoarchitecture of Zika Virus Infection in Human Neuroblastoma and Aedes Albopictus Cell Lines. Virology 2017, 501, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slonchak, A.; Hugo, L.E.; Freney, M.E.; Hall-Mendelin, S.; Amarilla, A.A.; Torres, F.J.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Hall, R.A.; et al. Zika Virus Noncoding RNA Suppresses Apoptosis and Is Required for Virus Transmission by Mosquitoes. Nat. Commun. 2020, 11, 2205. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J. Arboviruses and Apoptosis: The Role of Cell Death in Determining Vector Competence. J. Gen. Virol. 2016, 97, 1033–1036. [Google Scholar] [CrossRef]
- Medina-Magües, L.G.; Gergen, J.; Jasny, E.; Petsch, B.; Lopera-Madrid, J.; Medina-Magües, E.S.; Salas-Quinchucua, C.; Osorio, J.E. MRNA Vaccine Protects against Zika Virus. Vaccines 2021, 9, 1464. [Google Scholar] [CrossRef]
- Xie, X.; Zou, J.; Shan, C.; Shi, P.-Y. Small Molecules and Antibodies for Zika Therapy. J. Infect. Dis. 2017, 216, S945–S950. [Google Scholar] [CrossRef] [Green Version]
- Clain, E.; Sinigaglia, L.; Koishi, A.C.; Gorgette, O.; Gadea, G.; Viranaicken, W.; Krejbich-Trotot, P.; Mavingui, P.; Desprès, P.; Nunes Duarte dos Santos, C.; et al. Extract from Aphloia Theiformis, an Edible Indigenous Plant from Reunion Island, Impairs Zika Virus Attachment to the Host Cell Surface. Sci. Rep. 2018, 8, 10856. [Google Scholar] [CrossRef] [Green Version]
- Baz, M.; Boivin, G. Antiviral Agents in Development for Zika Virus Infections. Pharmaceuticals 2019, 12, 101. [Google Scholar] [CrossRef] [Green Version]
- Bernatchez, J.A.; Tran, L.T.; Li, J.; Luan, Y.; Siqueira-Neto, J.L.; Li, R. Drugs for the Treatment of Zika Virus Infection. J. Med. Chem. 2020, 63, 470–489. [Google Scholar] [CrossRef]
- Han, Y.; Mesplède, T. Investigational Drugs for the Treatment of Zika Virus Infection: A Preclinical and Clinical Update. Expert Opin. Investig. Drugs 2018, 27, 951–962. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, N.; Aarthy, M.; Singh, S.K.; Giri, R. Mechanistic Insights into Zika Virus NS3 Helicase Inhibition by Epigallocatechin-3-Gallate. ACS Omega 2020, 5, 11217–11226. [Google Scholar] [CrossRef] [PubMed]
- Saiz, J.-C.; de Oya, N.J.; Blázquez, A.-B.; Escribano-Romero, E.; Martín-Acebes, M.A. Host-Directed Antivirals: A Realistic Alternative to Fight Zika Virus. Viruses 2018, 10, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, J.; Allison, C.; Preston, S.; Pellegrini, M. Therapeutic Manipulation of Host Cell Death Pathways to Facilitate Clearance of Persistent Viral Infections. J. Leukoc. Biol. 2018, 103, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Bulanova, D.; Ianevski, A.; Bugai, A.; Akimov, Y.; Kuivanen, S.; Paavilainen, H.; Kakkola, L.; Nandania, J.; Turunen, L.; Ohman, T.; et al. Antiviral Properties of Chemical Inhibitors of Cellular Anti-Apoptotic Bcl-2 Proteins. Viruses 2017, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Rider, T.H.; Zook, C.E.; Boettcher, T.L.; Wick, S.T.; Pancoast, J.S.; Zusman, B.D. Broad-Spectrum Antiviral Therapeutics. PLoS ONE 2011, 6, e22572. [Google Scholar] [CrossRef] [Green Version]
- Kuivanen, S.; Bespalov, M.M.; Nandania, J.; Ianevski, A.; Velagapudi, V.; De Brabander, J.K.; Kainov, D.E.; Vapalahti, O. Obatoclax, Saliphenylhalamide and Gemcitabine Inhibit Zika Virus Infection in Vitro and Differentially Affect Cellular Signaling, Transcription and Metabolism. Antivir. Res. 2017, 139, 117–128. [Google Scholar] [CrossRef]
- Su, K.Y.; Balasubramaniam, V.R.M.T. Zika Virus as Oncolytic Therapy for Brain Cancer: Myth or Reality? Front. Microbiol. 2019, 10, 2715. [Google Scholar] [CrossRef] [Green Version]
- Lawler, S.E.; Speranza, M.-C.; Cho, C.-F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841. [Google Scholar] [CrossRef] [Green Version]
- Vandenabeele, P.; Vandecasteele, K.; Bachert, C.; Krysko, O.; Krysko, D.V. Immunogenic Apoptotic Cell Death and Anticancer Immunity. In Apoptosis in Cancer Pathogenesis and Anti-cancer Therapy; Gregory, C.D., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; Volume 930, pp. 133–149. ISBN 978-3-319-39404-6. [Google Scholar]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika Virus Has Oncolytic Activity against Glioblastoma Stem Cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wu, J.; Ye, Q.; Ma, F.; Zhu, Q.; Wu, Y.; Shan, C.; Xie, X.; Li, D.; Zhan, X.; et al. Treatment of Human Glioblastoma with a Live Attenuated Zika Virus Vaccine Candidate. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin Avβ5 Axis. Cell Stem Cell 2020, 26, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Mazzoccoli, L.; Jash, A.; Govero, J.; Bais, S.S.; Hu, T.; Fontes-Garfias, C.R.; Shan, C.; Okada, H.; Shresta, S.; et al. Zika Virus Oncolytic Activity Requires CD8+ T Cells and Is Boosted by Immune Checkpoint Blockade. JCI Insight 2020, 6, e144619. [Google Scholar] [CrossRef] [PubMed]
- Mazar, J.; Li, Y.; Rosado, A.; Phelan, P.; Kedarinath, K.; Parks, G.D.; Alexander, K.A.; Westmoreland, T.J. Zika Virus as an Oncolytic Treatment of Human Neuroblastoma Cells Requires CD24. PLoS ONE 2018, 13, e0200358. [Google Scholar] [CrossRef] [PubMed]
- Altevogt, P.; Sammar, M.; Hüser, L.; Kristiansen, G. Novel Insights into the Function of CD24: A Driving Force in Cancer. Int. J. Cancer 2021, 148, 546–559. [Google Scholar] [CrossRef]
- Zwernik, S.D.; Adams, B.H.; Raymond, D.A.; Warner, C.M.; Kassam, A.B.; Rovin, R.A.; Akhtar, P. AXL Receptor Is Required for Zika Virus Strain MR-766 Infection in Human Glioblastoma Cell Lines. Mol. Ther.-Oncolytics 2021, 23, 447–457. [Google Scholar] [CrossRef]
- Delaunay, T.; Nader, J.; Grard, M.; Farine, I.; Hedwig, V.; Foloppe, J.; Blondy, T.; Violland, M.; Pouliquen, D.; Grégoire, M.; et al. High Oncolytic Activity of a Double-Deleted Vaccinia Virus Copenhagen Strain against Malignant Pleural Mesothelioma. Mol. Ther.-Oncolytics 2020, 18, 573–578. [Google Scholar] [CrossRef]
- Delaunay, T.; Achard, C.; Boisgerault, N.; Grard, M.; Petithomme, T.; Chatelain, C.; Dutoit, S.; Blanquart, C.; Royer, P.-J.; Minvielle, S.; et al. Frequent Homozygous Deletions of Type I Interferon Genes in Pleural Mesothelioma Confer Sensitivity to Oncolytic Measles Virus. J. Thorac. Oncol. 2020, 15, 827–842. [Google Scholar] [CrossRef]
- Lemos de Matos, A.; Franco, L.S.; McFadden, G. Oncolytic Viruses and the Immune System: The Dynamic Duo. Mol. Ther.-Methods Clin. Dev. 2020, 17, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Kaid, C.; Madi, R.A.D.S.; Astray, R.; Goulart, E.; Caires-Junior, L.C.; Mitsugi, T.G.; Moreno, A.C.R.; Castro-Amarante, M.F.; Pereira, L.R.; Porchia, B.F.M.M.; et al. Safety, Tumor Reduction, and Clinical Impact of Zika Virus Injection in Dogs with Advanced-Stage Brain Tumors. Mol. Ther. 2020, 28, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Monel, B.; Compton, A.A.; Bruel, T.; Amraoui, S.; Burlaud-Gaillard, J.; Roy, N.; Guivel-Benhassine, F.; Porrot, F.; Génin, P.; Meertens, L.; et al. Zika Virus Induces Massive Cytoplasmic Vacuolization and Paraptosis-like Death in Infected Cells. EMBO J. 2017, 36, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Beys-da-Silva, W.O.; Quincozes-Santos, A.; Tureta, E.F.; Rosa, R.L.; Berger, M.; Bobermin, L.D.; Souza, D.O.; Guimarães, J.A.; Santi, L. Association between Zika Virus and Future Neurological Diseases. J. Neurol. Sci. 2020, 409, 116617. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turpin, J.; El Safadi, D.; Lebeau, G.; Krejbich, M.; Chatelain, C.; Desprès, P.; Viranaïcken, W.; Krejbich-Trotot, P. Apoptosis during ZIKA Virus Infection: Too Soon or Too Late? Int. J. Mol. Sci. 2022, 23, 1287. https://doi.org/10.3390/ijms23031287
Turpin J, El Safadi D, Lebeau G, Krejbich M, Chatelain C, Desprès P, Viranaïcken W, Krejbich-Trotot P. Apoptosis during ZIKA Virus Infection: Too Soon or Too Late? International Journal of Molecular Sciences. 2022; 23(3):1287. https://doi.org/10.3390/ijms23031287
Chicago/Turabian StyleTurpin, Jonathan, Daed El Safadi, Grégorie Lebeau, Morgane Krejbich, Camille Chatelain, Philippe Desprès, Wildriss Viranaïcken, and Pascale Krejbich-Trotot. 2022. "Apoptosis during ZIKA Virus Infection: Too Soon or Too Late?" International Journal of Molecular Sciences 23, no. 3: 1287. https://doi.org/10.3390/ijms23031287