
Transcriptional Organization of the Salmonella Typhimurium Phage P22 pid ORFan Locus

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
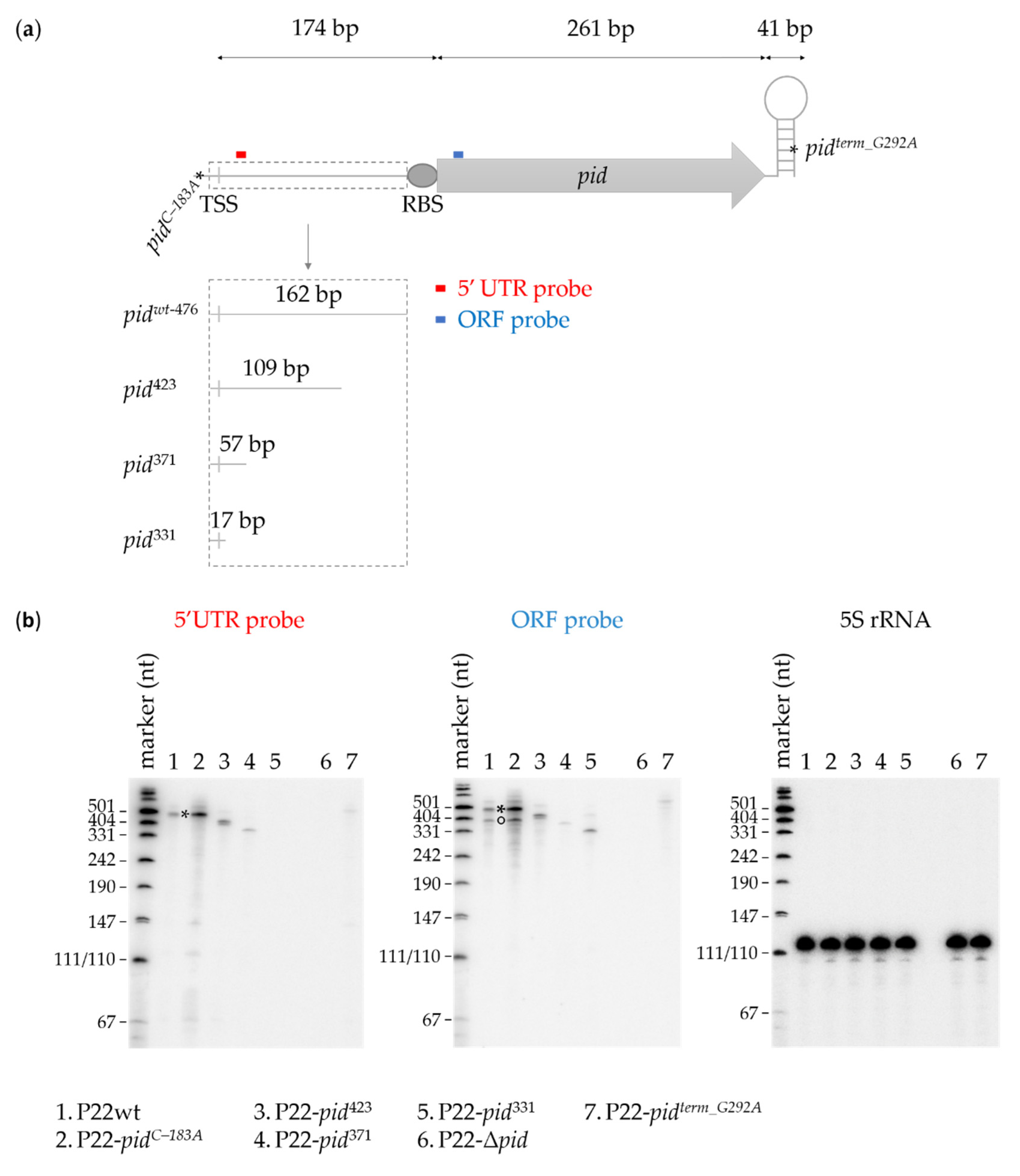
2. Results
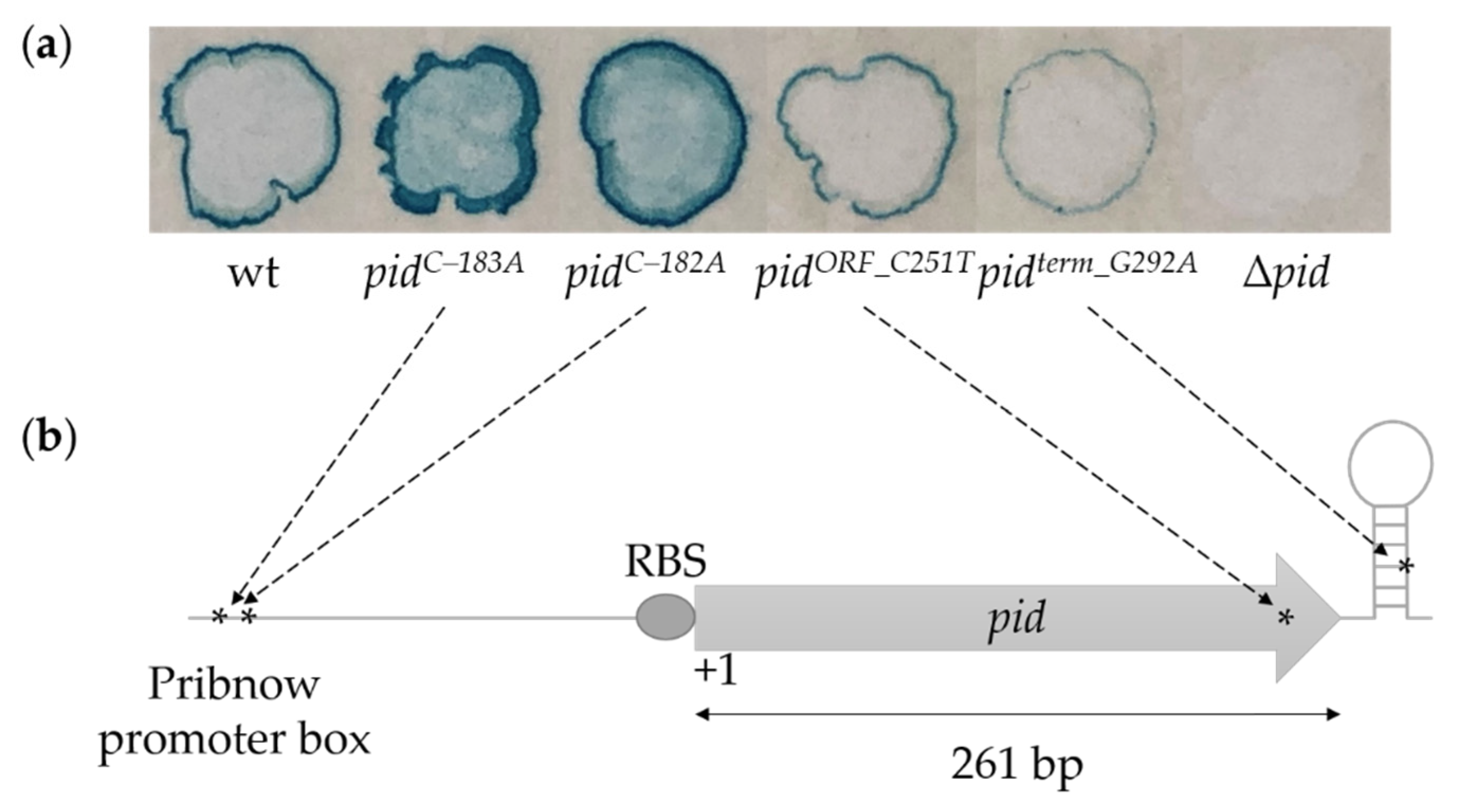
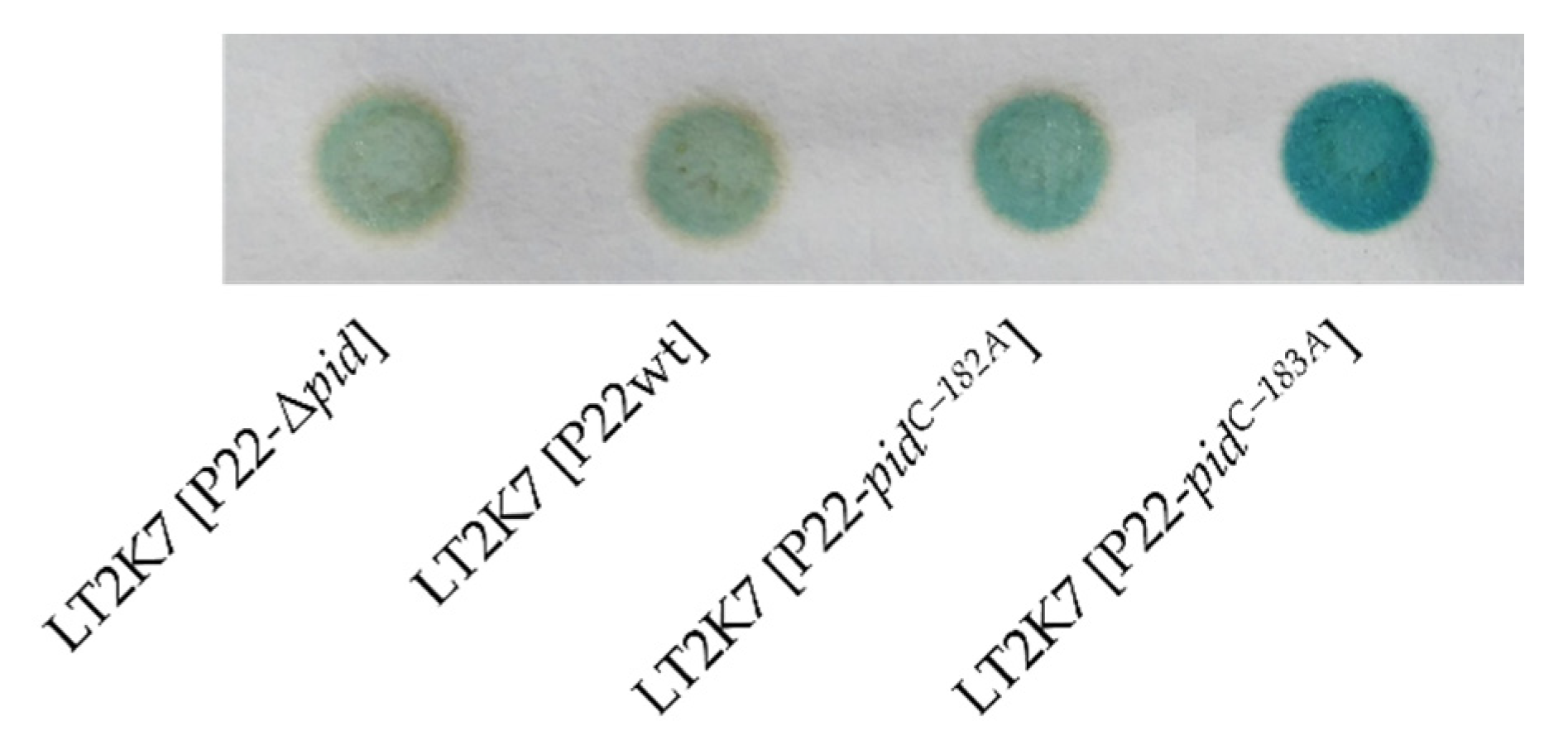
2.1. Genetic Screen for Functional Mutations in the P22 pid Locus
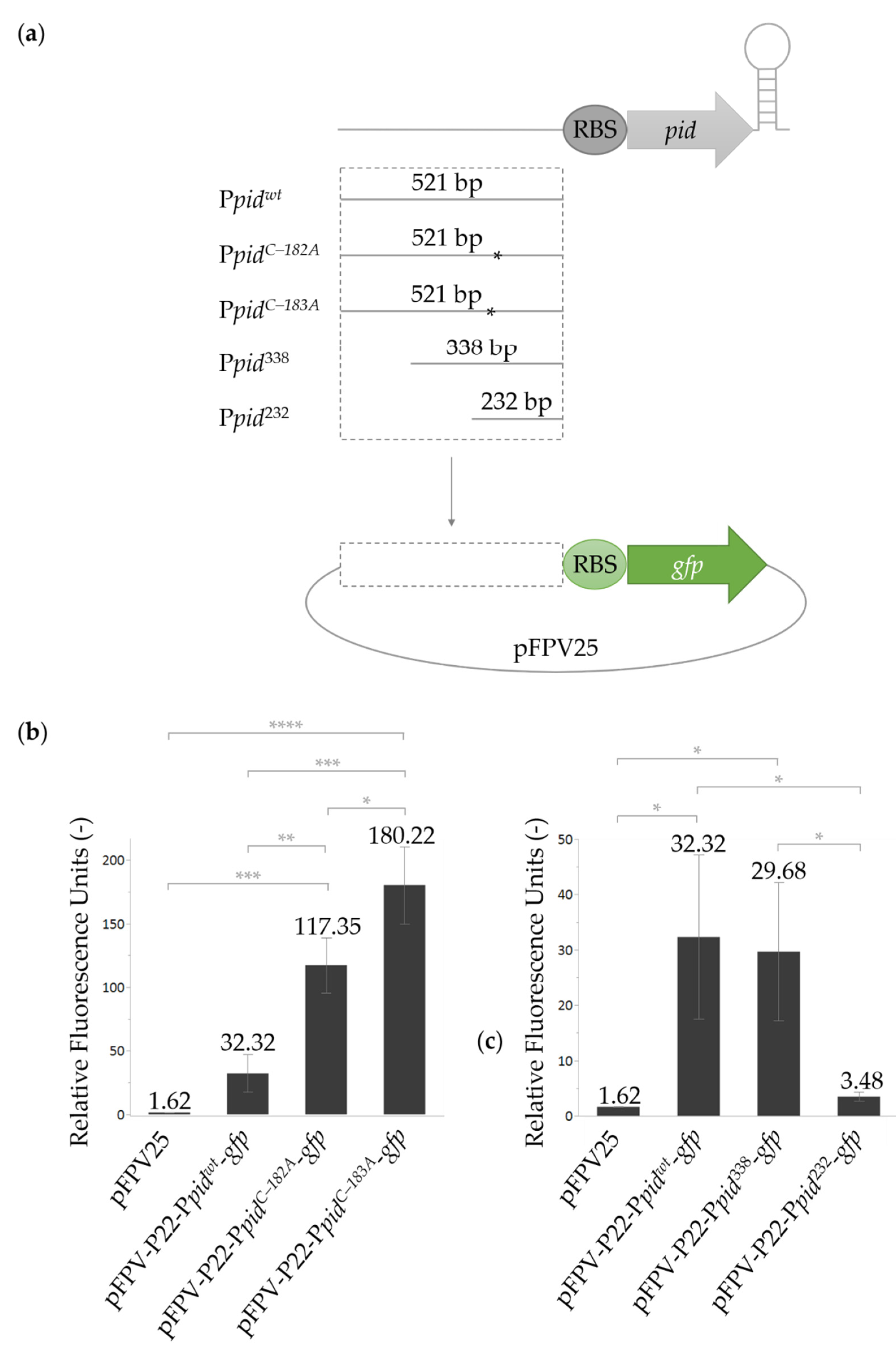
2.2. Characterization of the P22 pid Promoter
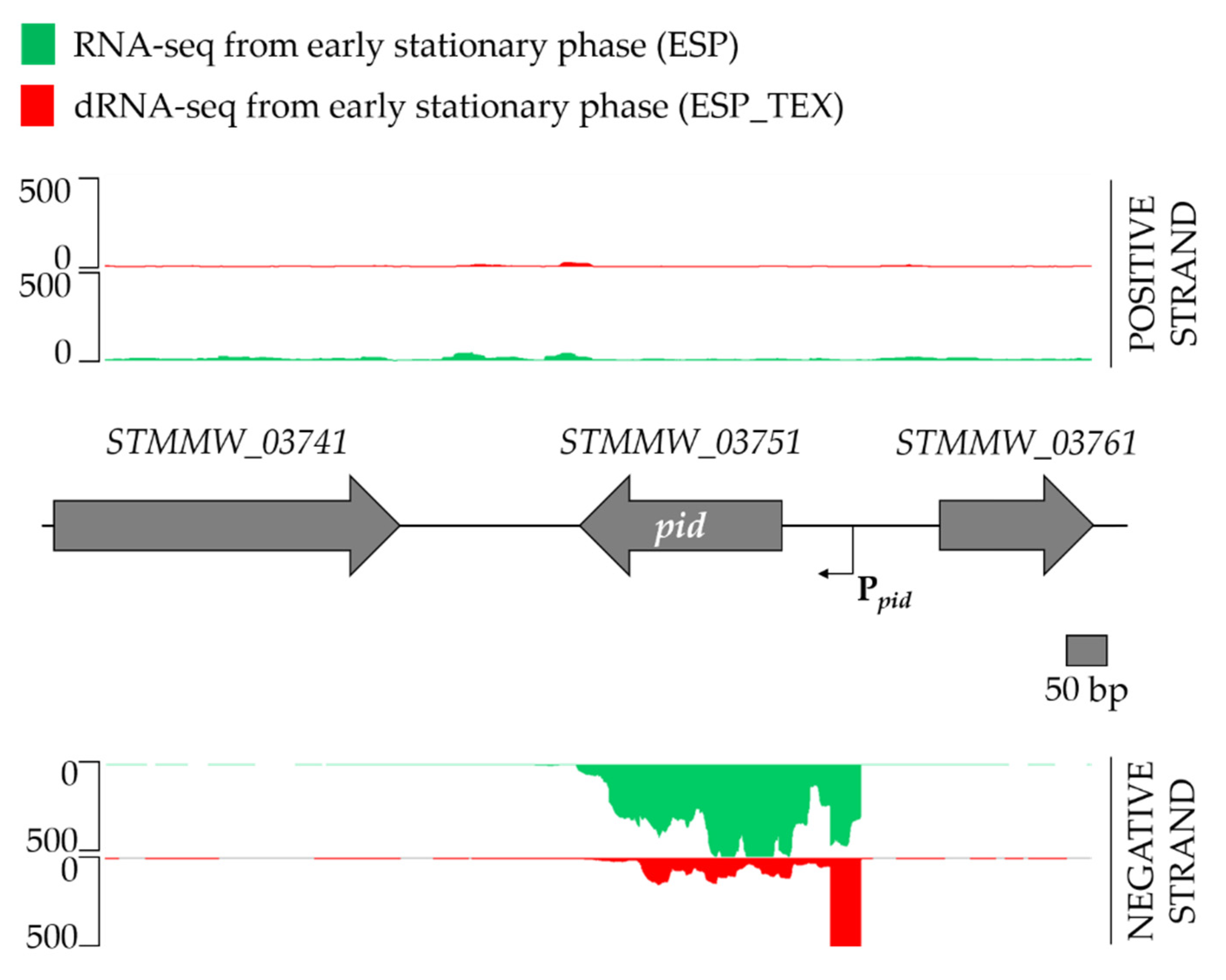
2.3. Characterization of the P22 pid Transcript
3. Discussion
4. Materials and Methods
4.1. Strains, Phages, and Growth Conditions
4.2. Construction of Lysogens
4.3. Construction of Phage Mutants
4.4. Construction of Plasmids
4.5. DES-Mutagenesis Screen
4.6. β-Galactosidase Assay
4.7. Fluorescence Population Level Measurement
4.8. Statistical Analysis
4.9. Northern Blotting
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Comeau, A.M.; Hatfull, G.F.; Krisch, H.M.; Lindell, D.; Mann, N.H.; Prangishvili, D. Exploring the prokaryotic virosphere. Res. Microbiol. 2008, 159, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Fischer, D. Identification and investigation of ORFans in the viral world. BMC Genom. 2008, 9, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipa-Silva, A.; Parreira, R.; Martínez-Puchol, S.; Bofill-Mas, S.; Crespo, M.T.B.; Nunes, M. The Unexplored Virome of Two Atlantic Coast Fish: Contribution of Next-Generation Sequencing to Fish Virology. Foods 2020, 9, 1634. [Google Scholar] [CrossRef]
- Owen, S.V.; Canals, R.; Wenner, N.; Hammarlöf, D.L.; Kröger, C.; Hinton, J.C.D. A window into lysogeny: Revealing temperate phage biology with transcriptomics. Microb. Genom. 2020, 6, e000330. [Google Scholar] [CrossRef]
- Ang, D.; Georgopoulos, C. An orfan no more: The bacteriophage T4 39.2 gene product, NWGI, modulates GroEL chaperone function. Genetics 2012, 190, 989–1000. [Google Scholar] [CrossRef] [Green Version]
- Boon, M.; De Zitter, E.; De Smet, J.; Wagemans, J.; Voet, M.; Pennemann, F.L.; Schalck, T.; Kuznedelov, K.; Severinov, K.; Van Meervelt, L.; et al. ‘Drc’, a structurally novel ssDNA-binding transcription regulator of N4-related bacterial viruses. Nucleic Acids Res. 2020, 48, 445–459. [Google Scholar] [CrossRef]
- Wagemans, J.; Blasdel, B.G.; Van den Bossche, A.; Uytterhoeven, B.; De Smet, J.; Paeshuyse, J.; Cenens, W.; Aertsen, A.; Uetz, P.; Delattre, A.S.; et al. Functional elucidation of antibacterial phage ORFans targeting Pseudomonas aeruginosa. Cell. Microbiol. 2014, 16, 1822–1835. [Google Scholar] [CrossRef]
- Mattenberger, Y.; Silva, F.; Belin, D. 55.2 a phage T4 ORFan gene, encodes an inhibitor of Escherichia coli topoisomerase I, increases phage fitness. PLoS ONE 2015, 10, e0124309. [Google Scholar] [CrossRef] [Green Version]
- Cenens, W.; Makumi, A.; Govers, S.K.; Lavigne, R.; Aertsen, A. Viral Transmission Dynamics at Single-Cell Resolution Reveal Transiently Immune Subpopulations Caused by a Carrier State Association. PLoS Genet. 2015, 11, e1005770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenens, W.; Mebrhatu, M.T.; Makumi, A.; Ceyssens, P.J.; Lavigne, R.; Van Houdt, R.; Taddei, F.; Aertsen, A. Expression of a Novel P22 ORFan Gene Reveals the Phage Carrier State in Salmonella Typhimurium. PLoS Genet. 2013, 9, e1003269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, H.; Aiba, H. Differential contributions of two elements of rho-independent terminator to transcription termination and mRNA stabilization. Biochimie 1996, 78, 1035–1042. [Google Scholar] [CrossRef]
- Wilson, K.S.; von Hippel, P.H. Transcription termination at intrinsic terminators: The role of the RNA hairpin. Proc. Natl. Acad. Sci. USA 1995, 92, 8793–8797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Coppens, L.; Lavigne, R. SAPPHIRE: A neural network based classifier for σ70 promoter prediction in Pseudomonas. BMC Bioinform. 2020, 21, 415. [Google Scholar] [CrossRef]
- Valdivia, R.H.; Falkow, S. Fluorescence-based isolation of bacterial genes expressed within host cells. Science 1997, 277, 2007–2011. [Google Scholar] [CrossRef] [Green Version]
- Fournier, B.; Lu, C.Y.; Lagrange, P.H.; Krishnamoorthy, R.; Philippon, A. Point mutation in the Pribnow box, the molecular basis of β-lactamase overproduction in Klebsiella oxytoca. Antimicrob. Agents Chemother. 1995, 39, 1365–1368. [Google Scholar] [CrossRef] [Green Version]
- Chapon, C. Expression of malT, the regulator gene of the maltose region in Escherichia coli, is limited both at transcription and translation. EMBO J. 1982, 1, 369–374. [Google Scholar] [CrossRef]
- Kingsley, R.A.; Msefula, C.L.; Thomson, N.R.; Kariuki, S.; Holt, K.E.; Gordon, M.A.; Harris, D.; Clarke, L.; Whitehead, S.; Sangal, V.; et al. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 2009, 19, 2279–2287. [Google Scholar] [CrossRef] [Green Version]
- Hammarlöf, D.L.; Kröger, C.; Owen, S.V.; Canals, R.; Lacharme-Lora, L.; Wenner, N.; Schager, A.E.; Wells, T.J.; Henderson, I.R.; Wigley, P.; et al. Role of a single noncoding nucleotide in the evolution of an epidemic African clade of Salmonella. Proc. Natl. Acad. Sci. USA 2018, 115, E2614–E2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canals, R.; Hammarlöf, D.L.; Kröger, C.; Owen, S.V.; Fong, W.Y.; Lacharme-Lora, L.; Zhu, X.; Wenner, N.; Carden, S.E.; Honeycutt, J.; et al. Adding function to the genome of African Salmonella Typhimurium ST313 strain D23580. PLoS Biol. 2019, 17, e3000059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.H.; Emory, S.A.; Bricker, A.L.; Bouvet, P.; Belasco, J.G. Structure and function of a bacterial mRNA stabilizer: Analysis of the 5′ untranslated region of ompA mRNA. J. Bacteriol. 1991, 173, 4578–4586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, T.E.; Yu, J.; Belasco, J.G. mRNA stabilization by the ompA 5′ untranslated region: Two protective elements hinder distinct pathways for mRNA degradation. RNA 1998, 4, 319–330. [Google Scholar] [PubMed]
- Cao, Y.; Li, J.; Jiang, N.; Dong, X. Mechanism for stabilizing mRNAs involved in methanol-dependent methanogenesis of cold-adaptive Methanosarcina mazei zm-15. Appl. Environ. Microbiol. 2014, 80, 1291–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Mao, X.H. The 5′ untranslated region of fruA mRNA in Myxococcus xanthus positively regulates the expression of its own gene. Chin. Sci. Bull. 2004, 49, 2266–2271. [Google Scholar] [CrossRef]
- Jian, H.; Xiong, L.; Xu, G.; Xiao, X.; Wang, F. Long 5′ untranslated regions regulate the RNA stability of the deep-sea filamentous phage SW1. Sci. Rep. 2016, 6, 21908. [Google Scholar] [CrossRef] [Green Version]
- Belin, D.; Mudd, E.A.; Prentki, P.; Yi-Yi, Y.; Krisch, H.M. Sense and antisense transcription of bacteriophage T4 gene 32. Processing and stability of the mRNAs. J. Mol. Biol. 1987, 194, 231–243. [Google Scholar] [CrossRef]
- Lioliou, E.; Sharma, C.M.; Caldelari, I.; Helfer, A.C.; Fechter, P.; Vandenesch, F.; Vogel, J.; Romby, P. Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet. 2012, 8, e1002782. [Google Scholar] [CrossRef]
- Obana, N.; Shirahama, Y.; Abe, K.; Nakamura, K. Stabilization of Clostridium perfringens collagenase mRNA by VR-RNA-dependent cleavage in 5′ leader sequence. Mol. Microbiol. 2010, 77, 1416–1428. [Google Scholar] [CrossRef]
- Martínez-Trujillo, M.; Sanchez-Trujillo, A.; Ceja, V.; Ávila-Moreno, F. Sequences required for transcription termination at the intrinsic λtI terminator. Can. J. Microbiol. 2010, 56, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Cisneros, B.; Court, D.; Sanchez, A.; Montañez, C. Point mutations in a transcription terminator, λtI, that affect both transcription termination and RNA stability. Gene 1996, 181, 127–133. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning (a Laboratory Manual); Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- McClelland, M.; Sanderson, K.E.; Spieth, J.; Clifton, S.W.; Latreille, P.; Courtney, L.; Porwollik, S.; Ali, J.; Dante, M.; Du, F.; et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 2001, 413, 852–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.T.; Thomason, L.C.; Sawitzke, J.A.; Costantino, N.; Court, D.L. Positive and negative selection using the tetA-sacB cassette: Recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res. 2013, 41, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherepanov, P.P.; Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 1995, 158, 9–14. [Google Scholar] [CrossRef]
- Davis, R.; Botstein, D.; Roth, J. Advanced Bacterial Genetics; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1980. [Google Scholar]
- Clark, D.J.; Maaløe, O. DNA replication and the division cycle in Escherichia coli. J. Mol. Biol. 1967, 23, 99–112. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Characteristic | Source or Reference |
|---|---|---|
| Strains | ||
| LT2 | Salmonella Typhimurium wild-type | [35] |
| LT2K7 | LT2 dgoT::MudK$Insertion of MudK in the dgo operon, resulting in a translational lacZ reporter fusion to the dgoT gene | [12] |
| LT2 [P22wt] | LT2 lysogenized with P22wt | This study |
| LT2K7 [P22wt] | LT2K7 lysogenized with P22wt | This study |
| LT2K7 [P22-Δpid] | LT2K7 lysogenized with P22-Δpid | This study |
| LT2K7 [P22-pidC–183A] | LT2K7 lysogenized with P22-pidC–183A | This study |
| LT2K7 [P22-pidC–182A] | LT2K7 lysogenized with P22-pidC–182A | This study |
| D23580 | Salmonella Typhimurium strain, naturally lysogenized with BTP1 prophage | [20] |
| E. coli XTL298 | Contains the tetA-sacB cassette | [36] |
| Bacteriophages | ||
| P22wt | Wild-type P22 phage | Salmonella Genetic Stock Centre (SGSC) 1 |
| P22-Δpid | P22 with a deletion of the pid ORF and regulatory region | This study |
| P22-pidORF_C251T | P22 with a C-to-T point mutation, 251 bp downstream of the pid start codon | This study |
| P22-pidterm_G292A | P22 with a G-to-A point mutation in the rho-independent terminator, 292 bp downstream of the pid start codon | This study |
| P22-pidC–183A | P22 with a C-to-A point mutation, 183 bp upstream of the pid start codon | This study |
| P22-pidC–182A | P22 with a C-to-A point mutation, 182 bp upstream of the pid start codon | This study |
| P22-pid423 | P22 with a 3′ truncated 5′UTR region of 109 bp, followed by an intact pid RBS | This study |
| P22-pid371 | P22 with a 3′ truncated 5′UTR region of 57 bp, followed by an intact pid RBS | This study |
| P22-pid331 | P22 with a 3′ truncated 5′UTR region of 17 bp, followed by an intact pid RBS | This study |
| Plasmids | ||
| pKD46 | Encodes λ-red recombineering genes under control of L-arabinose inducible promoter | [37] |
| pKD13 | Harbors frt-kan-frt site for construction of deletions by recombineering | [37] |
| pCP20 | Encodes flippase (FLP) for recombining frt sites | [38] |
| pFPV25 | Encodes promoterless GFP | [17] |
| pFPV-P22-Ppidwt-gfp | Contains 521 bp of the 5′ regulatory region of P22wt upstream of gfp in the pFPV25 plasmid | This study |
| pFPV-P22-PpidC–182A-gfp | Contains 521 bp of the 5′ regulatory region of P22-PpidC–182A upstream of gfp in the pFPV25 plasmid | This study |
| pFPV-P22-PpidC–183A-gfp | Contains 521 bp of the 5′ regulatory region of P22-PpidC–183A upstream of gfp in the pFPV25 plasmid | This study |
| pFPV-P22-Ppid338-gfp | Contains 338 bp of the 5′ regulatory region of P22wt upstream of gfp in the pFPV25 plasmid | This study |
| pFPV-P22-Ppid232-gfp | Contains 232 bp of the 5′ regulatory region of P22wt upstream of gfp in the pFPV25 plasmid | This study |
| Primer Name | Sequence (5′–3′) 1 |
|---|---|
| P22_Δpid_Fw | GTGATGATGCCGAGCACGCCCATCTGGACTATCTCAACTAGTCGATTCAT ATTCCGGGGATCCGTCGACC |
| P22_Δpid_Rev | CTTATACCATCGACTGGATATTATTCGTTTTATCCCGTCTATGTGGGGGGGGGGATAAAA TGTAGGCTGGAGCTGCTTCG |
| P22_pid_Fw | GTCAGGATCC ACAGGTCTAACGCTTCCC |
| P22_pid_Rev | GATGTCTAGA GCATAAAGTTTCTTGTGGTTG |
| P22_pid338_Fw | GTCAGGATCC ACTGGAATTTCTGTTCTTCAGTCA |
| P22_pid232_Fw | GTCAGGATCC TCATGACATGTGTCACATTTATA |
| P22_5UTRpid_tetAsacB_Fw | ATATCTTCAAGGTGGGCAATTTTTTGCTCTATATCTGACATGTCCACTCCTTT TCCTAATTTTTGTTGACACTCTATC |
| P22_5UTRpid_tetAsacB_Revv | ATTGCTTTAAGTTTACAGAACAATAATCCTTGGCTGGACGTAAGGTTTTGACA ATCAAAGGGAAAACTGTCCATATGC |
| P22_5UTR423pid_Fw | ATATCTTCAAGGTGGGCAATTTTTTGCTCTATATCTGACATGTCCACTCCTTT TCTGTCATGAGTACCTCATCG |
| P22_5UTR371pid_Fw | ATATCTTCAAGGTGGGCAATTTTTTGCTCTATATCTGACATGTCCACTCCTTT GGAGATATCTAAGATTGC |
| P22_5UTR331pid_Fw | ATATCTTCAAGGTGGGCAATTTTTTGCTCTATATCTGACATGTCCACTCCTTT TGTCAAAACCTTACGTCCAGCCAA |
| P225UTRpid_Rev | GATTCATGACATGTGTCACATTTATACCAACCAGATCATTGCTTTAAGTTTAC |
| Sal_5S_rRNA | TGGGACCACCGCGCTAGTGCCG |
| Pid_5UTR | ATCTAAGATTGCTATCACACTGC |
| Pid_ORF | GATAATATCTTCAAGGTGGGCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolput, S.; Makumi, A.; Wicke, L.; Bäcker, L.E.; Cenens, W.; Briers, Y.; Wenner, N.A.; Owen, S.V.; Hinton, J.C.D.; Lavigne, R.; et al. Transcriptional Organization of the Salmonella Typhimurium Phage P22 pid ORFan Locus. Int. J. Mol. Sci. 2022, 23, 1253. https://doi.org/10.3390/ijms23031253
Wolput S, Makumi A, Wicke L, Bäcker LE, Cenens W, Briers Y, Wenner NA, Owen SV, Hinton JCD, Lavigne R, et al. Transcriptional Organization of the Salmonella Typhimurium Phage P22 pid ORFan Locus. International Journal of Molecular Sciences. 2022; 23(3):1253. https://doi.org/10.3390/ijms23031253
Chicago/Turabian StyleWolput, Sanne, Angela Makumi, Laura Wicke, Leonard E. Bäcker, William Cenens, Yves Briers, Nicolas A. Wenner, Siân V. Owen, Jay C. D. Hinton, Rob Lavigne, and et al. 2022. "Transcriptional Organization of the Salmonella Typhimurium Phage P22 pid ORFan Locus" International Journal of Molecular Sciences 23, no. 3: 1253. https://doi.org/10.3390/ijms23031253