
Effect of Novel AKT Inhibitor Vevorisertib as Single Agent and in Combination with Sorafenib on Hepatocellular Carcinoma in a Cirrhotic Rat Model
 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. In Vitro Experiments
2.2. In Vivo Experiments
2.2.1. Safety Assessment
2.2.2. Effect on Tumor Progression
2.2.3. Effect on Tumor Cell Proliferation
2.2.4. Effect of Treatment on Tumor Vascularization and Liver Fibrosis
3. Discussion
4. Materials and Methods
4.1. In Vitro Experiments
4.2. Treatment
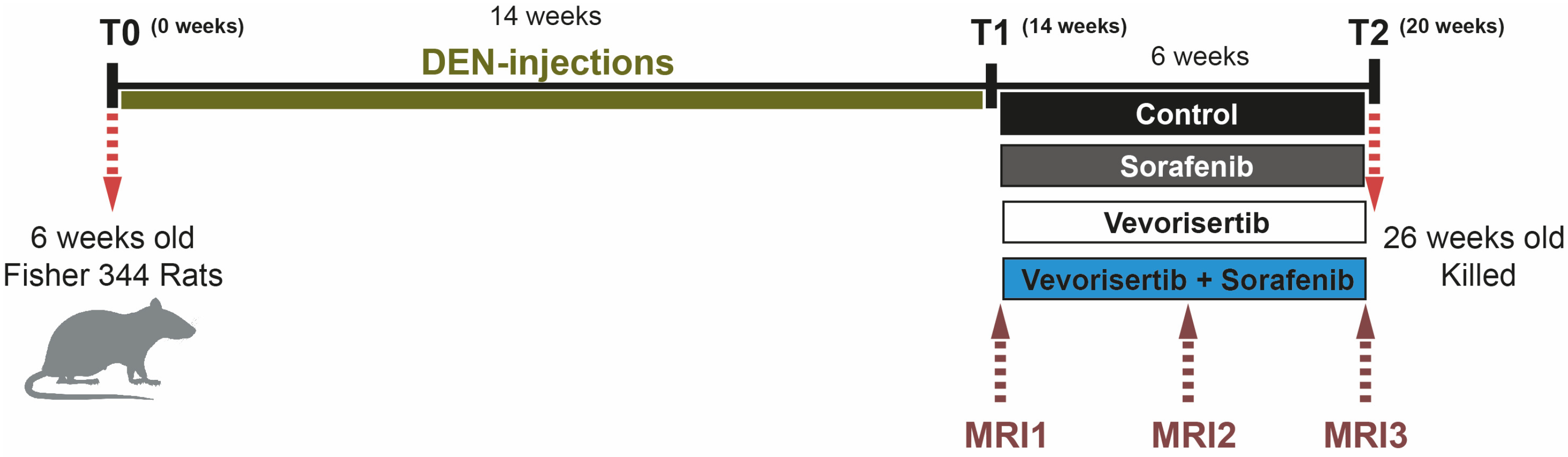
4.3. Animals and Treatment Protocol
4.4. MRI Analyses
4.5. Morphological and Histopathological Analyses
4.6. Immunohistochemical and Immunofluorescence Analyses
4.7. Real Time Polymerase Chain Reaction (qPCR)
4.8. Immunoblot Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Macek Jilkova, Z.; Ghelfi, J.; Decaens, T. Immunomodulation for hepatocellular carcinoma therapy: Current challenges. Curr. Opin. Oncol. 2022, 34, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Garrett, M.D.; Walton, M.I.; Raynaud, F.; de Bono, J.S.; Workman, P. Targeting the PI3K-AKT-mTOR pathway: Progress, pitfalls, and promises. Curr. Opin. Pharmacol. 2008, 8, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT--a major therapeutic target. Biochim. Et Biophys. Acta 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology (Baltim. Md.) 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. (Northwood Lond. Engl.) 2010, 27, 255–261. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Q.; Liu, J.; Cao, H. Inhibition of the PI3K/Akt signaling pathway reverses sorafenib-derived chemo-resistance in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 9377–9384. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Macias, R.I.R.; Monte, M.J.; Romero, M.R.; Asensio, M.; Sanchez-Martin, A.; Cives-Losada, C.; Temprano, A.G.; Espinosa-Escudero, R.; Reviejo, M.; et al. Molecular Bases of Drug Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1663. [Google Scholar] [CrossRef]
- Shi, C.; Kwong, D.L.-W.; Li, X.; Wang, X.; Fang, X.; Sun, L.; Tang, Y.; Guan, X.-Y.; Li, S.-S. MAEL Augments Cancer Stemness Properties and Resistance to Sorafenib in Hepatocellular Carcinoma through the PTGS2/AKT/STAT3 Axis. Cancers 2022, 14, 2880. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.P.; Xiong, B.H.; Zhang, Y.X.; Wang, S.L.; Zuo, Q.; Li, J. FXYD5 promotes sorafenib resistance through the Akt/mTOR signaling pathway in hepatocellular carcinoma. Eur. J. Pharmacol. 2022, 931, 175186. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.J.; Wankell, M.; Palamuthusingam, P.; McFarlane, C.; Hebbard, L. Targeting the PI3K/Akt/mTOR Pathway in Hepatocellular Carcinoma. Biomedicines 2021, 9, 1639. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Savage, R.E.; Eathiraj, S.; Meade, J.; Wick, M.J.; Hall, T.; Abbadessa, G.; Schwartz, B. Targeting AKT1-E17K and the PI3K/AKT Pathway with an Allosteric AKT Inhibitor, ARQ 092. PLoS ONE 2015, 10, e0140479. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Kuyucu, A.Z.; Kurma, K.; Ahmad Pour, S.T.; Roth, G.S.; Abbadessa, G.; Yu, Y.; Schwartz, B.; Sturm, N.; Marche, P.N.; et al. Combination of AKT inhibitor ARQ 092 and sorafenib potentiates inhibition of tumor progression in cirrhotic rat model of hepatocellular carcinoma. Oncotarget 2018, 9, 11145–11158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, G.S.; Macek Jilkova, Z.; Zeybek Kuyucu, A.; Kurma, K.; Ahmad Pour, S.T.; Abbadessa, G.; Yu, Y.; Busser, B.; Marche, P.N.; Leroy, V.; et al. Efficacy of AKT Inhibitor ARQ 092 Compared with Sorafenib in a Cirrhotic Rat Model with Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 2157–2165. [Google Scholar] [CrossRef] [Green Version]
- Kurma, K.; Manches, O.; Chuffart, F.; Sturm, N.; Gharzeddine, K.; Zhang, J.; Mercey-Ressejac, M.; Rousseaux, S.; Millet, A.; Lerat, H.; et al. DEN-Induced Rat Model Reproduces Key Features of Human Hepatocellular Carcinoma. Cancers 2021, 13, 4981. [Google Scholar] [CrossRef]
- Kostaras, E.; Kaserer, T.; Lazaro, G.; Heuss, S.F.; Hussain, A.; Casado, P.; Hayes, A.; Yandim, C.; Palaskas, N.; Yu, Y.; et al. A systematic molecular and pharmacologic evaluation of AKT inhibitors reveals new insight into their biological activity. Br. J. Cancer 2020, 123, 542–555. [Google Scholar] [CrossRef]
- Yang, C.J.; Wu, M.H.; Tsai, J.J.; Hsu, F.T.; Hsia, T.C.; Liu, K.C.; Kuo, Y.C. Inactivation of AKT/ERK Signaling and Induction of Apoptosis Are Associated With Amentoflavone Sensitization of Hepatocellular Carcinoma to Lenvatinib. Anticancer Res. 2022, 42, 2495–2505. [Google Scholar] [CrossRef]
- Mroweh, M.; Roth, G.; Decaens, T.; Marche, P.N.; Lerat, H.; Macek Jilkova, Z. Targeting Akt in Hepatocellular Carcinoma and Its Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 1794. [Google Scholar] [CrossRef]
- Thabut, D.; Shah, V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: New targets for the treatment of portal hypertension? J. Hepatol. 2010, 53, 976–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, K.; Huang, Z.; Lin, T.; Liu, S.; Chang, H.; Yan, Z.; Zhang, H.; Liu, C. New Insight into the Anti-liver Fibrosis Effect of Multitargeted Tyrosine Kinase Inhibitors: From Molecular Target to Clinical Trials. Front. Pharmacol. 2015, 6, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gungor, M.Z.; Uysal, M.; Senturk, S. The Bright and the Dark Side of TGF-beta Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications. Cancers 2022, 14, 940. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, S.U.; Li, Y.H.; Roy, J.; Zhang, F.; Puchulu-Campanella, E.; Lindeman, S.D.; Srinivasarao, M.; Tsoyi, K.; Liang, X.; Ayaub, E.A.; et al. Targeted inhibition of PI3 kinase/mTOR specifically in fibrotic lung fibroblasts suppresses pulmonary fibrosis in experimental models. Sci. Transl. Med. 2020, 12, eaay3724. [Google Scholar] [CrossRef]
- Kunter, I.; Erdal, E.; Nart, D.; Yilmaz, F.; Karademir, S.; Sagol, O.; Atabey, N. Active form of AKT controls cell proliferation and response to apoptosis in hepatocellular carcinoma. Oncol. Rep. 2014, 31, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Carreres, L.; Mercey-Ressejac, M.; Kurma, K.; Ghelfi, J.; Fournier, C.; Manches, O.; Chuffart, F.; Rousseaux, S.; Minoves, M.; Decaens, T.; et al. Chronic Intermittent Hypoxia Increases Cell Proliferation in Hepatocellular Carcinoma. Cells 2022, 11, 2051. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Sorafenib | Vevorisertib | Vevorisertib + Soraf | p-Value | |
|---|---|---|---|---|---|
| Body Weight (g) | 291 ± 5.0 | 290 ± 4.1 | 290 ± 3.3 | 281 ± 3.3 | 0.1520 |
| Liver | |||||
| Weight (g) | 13.0 ± 0.4 | 13.1 ± 0.3 | 13.1 ± 0.4 | 11.6 ± 0.6 | 0.0567 |
| TG (g/L) | 31.5 ± 4.9 | 26.6 ± 1.9 | 28.5 ± 2.5 | 26.3 ± 1.7 | 0.5783 |
| Blood | |||||
| Albumin (g/dL) | 3.5 ± 0.18 | 3.6 ± 0.06 | 3.6 ± 0.03 | 3.3 ±0.05 *, # | 0.0127 |
| AST (U/L) | 97.4 ± 5.9 | 90.2 ± 4.8 | 82.9 ± 2.9 | 75.3 ± 3.6 * | 0.0283 |
| ALT (U/L) | 69.3 ± 3.9 | 71.2 ± 3.4 | 61.8 ± 3.4 | 50.3 ± 3.0 *, ## | 0.0029 |
| ALP (U/L) | 199.2 ± 9.7 | 212.0 ± 7.6 | 184.6 ± 7.1 | 192.0 ± 5.7 | 0.1499 |
| GGT (U/L) | 14.0 ± 3.0 | 15.3 ± 2.1 | 7.7 ± 1.7 | 2.7 ± 1.1 *, ## | 0.0013 |
| PT (s) | 18.1 ± 0.5 | 19.2 ± 1.3 | 18.0 ± 0.3 | 18.5 ± 0.5 | 0.9013 |
| Total Bilirubin (mg/dL) | 0.25 ± 0.04 | 0.18 ± 0.01 | 0.15 ± 0.01 * | 0.14 ± 0.01 *** | 0.0002 |
| Creatinine (mg/dL) | 0.37 ± 0.02 | 0.33 ± 0.02 | 0.40 ± 0.01 | 0.31 ± 0.01 | 0.0700 |
| GLU (mg/dL) | 132.3 ± 7.8 | 141.1 ± 5.7 | 152.6 ± 7.0 | 140.0 ± 6.2 | 0.2618 |
| Cholesterol (mg/dL) | 84.4 ± 4.3 | 86.2 ± 3.0 | 102.6 ± 2.0 *, & | 84.7 ± 2.2 | 0.0044 |
| TG (g/L) | 78.4 ± 11.1 | 70.3 ± 10.7 | 54.0 ± 11.5 | 60.6 ± 13.1 | 0.4764 |
| Gene | Reverse Sequence (5′–3′) | Forward Sequence (5′–3′) |
|---|---|---|
| α-SMA | CATCTCCAGAGTCCAGCACA | ACTGGGACGACATGGAAAAG |
| COL1 | CTTCTGGGCAGAAAGGACAG | GCCAAGAAGACATCCCTGAA |
| TGF-β1 | TGGGACTGATCCCATTGATT | ATACGCCTGAGTAGCTGTCT |
| TIMP1 | TGGCTGAACAGGGAAACACT | CAGCAAAAGGCCTTCGTAAA |
| MMP2 | GGGTTTCTTCTGGCTCAGG | TCTGGCTATCCACAAGACTGG |
| MMP9 | GGAAAAGGAAGGAGGGTACG | CCACTCAGGGCCTTCAGAC |
| GAPDH | TTCAGCTCTGGGATGACCTT | CTCATGACCACAGTCCATGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurma, K.; Zeybek Kuyucu, A.; Roth, G.S.; Sturm, N.; Mercey-Ressejac, M.; Abbadessa, G.; Yu, Y.; Lerat, H.; Marche, P.N.; Decaens, T.; et al. Effect of Novel AKT Inhibitor Vevorisertib as Single Agent and in Combination with Sorafenib on Hepatocellular Carcinoma in a Cirrhotic Rat Model. Int. J. Mol. Sci. 2022, 23, 16206. https://doi.org/10.3390/ijms232416206
Kurma K, Zeybek Kuyucu A, Roth GS, Sturm N, Mercey-Ressejac M, Abbadessa G, Yu Y, Lerat H, Marche PN, Decaens T, et al. Effect of Novel AKT Inhibitor Vevorisertib as Single Agent and in Combination with Sorafenib on Hepatocellular Carcinoma in a Cirrhotic Rat Model. International Journal of Molecular Sciences. 2022; 23(24):16206. https://doi.org/10.3390/ijms232416206
Chicago/Turabian StyleKurma, Keerthi, Ayca Zeybek Kuyucu, Gaël S. Roth, Nathalie Sturm, Marion Mercey-Ressejac, Giovanni Abbadessa, Yi Yu, Herve Lerat, Patrice N. Marche, Thomas Decaens, and et al. 2022. "Effect of Novel AKT Inhibitor Vevorisertib as Single Agent and in Combination with Sorafenib on Hepatocellular Carcinoma in a Cirrhotic Rat Model" International Journal of Molecular Sciences 23, no. 24: 16206. https://doi.org/10.3390/ijms232416206