
Protein Kinase Inhibitors as a New Target for Immune System Modulation and Brain Cancer Management
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Basis on the Existing Relationship between the Immune System and Varied Brain Tumors
3. Protein Kinase Involvement as the Main Players in the Progression and Development of Malignant Brain Tumors: Overview on Astrocytoma and Glioblastoma
4. Strategies for Targeting Malignant Brain Tumors with Kinase Inhibitors: VEGF, HER2, CSF-1R, PI3K, ERKI/II Targeting
4.1. Angiogenic Inhibition in High-Grade Gliomas by Targeting VEGFR
4.2. Pharmacological Inhibition of HER2 in High-Grade Gliomas
4.3. CSF-1R Kinase Inhibitors Reduce Glioma Progression through TAMs Depletion
4.4. Activity of Novel PI3K Inhibitors in Vivo and in Vitro Glioma Models
4.5. ERK Inhibition Suppresses Glioma Growth
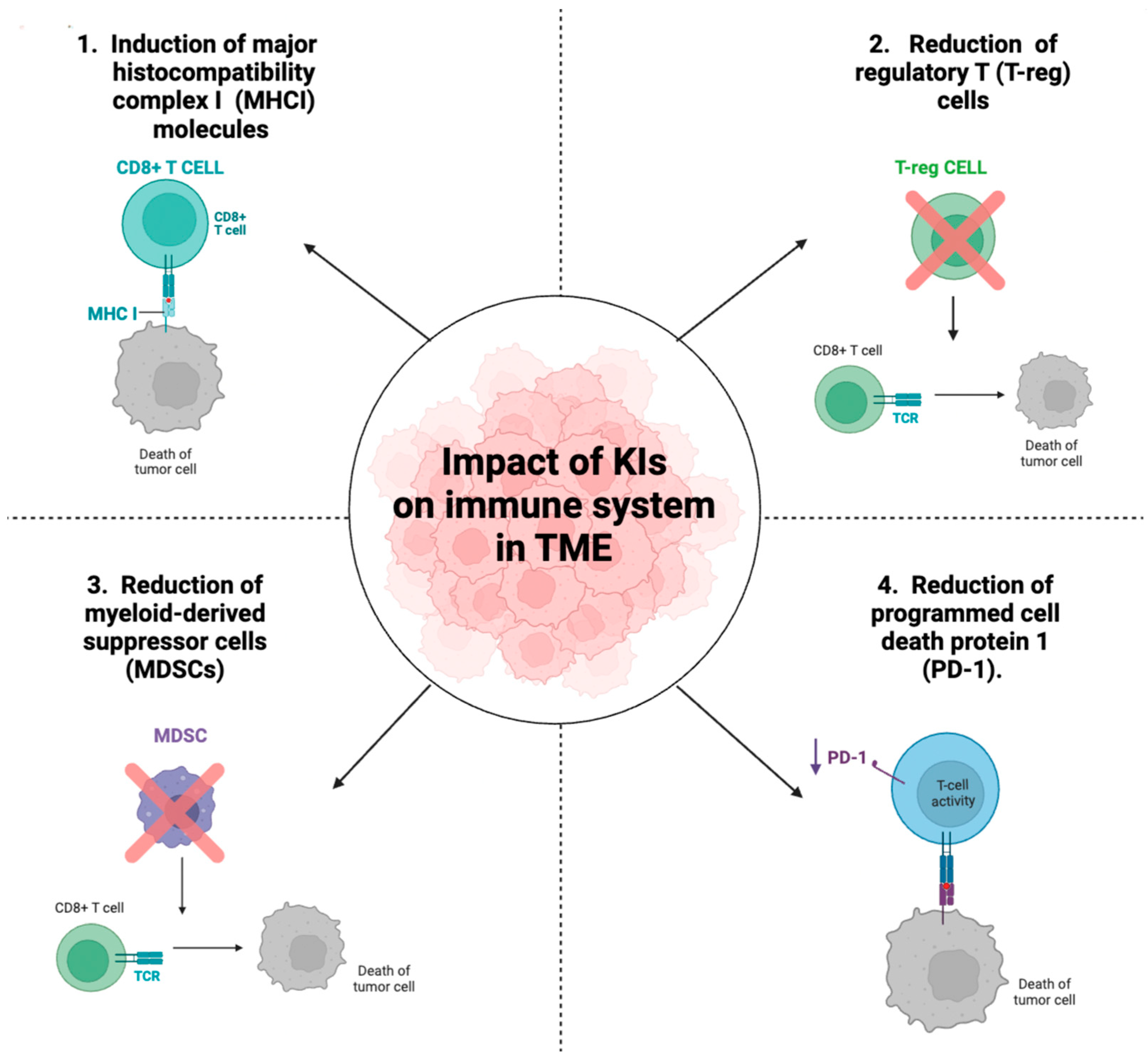
5. Impact of Kinase Inhibitors on Immune System
6. Side Effects and Resistance Phenomena of Current KIs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Chen, B.; Liang, T.; Yang, P.; Wang, H.; Liu, Y.; Yang, F.; You, G. Classifying lower grade glioma cases according to whole genome gene expression. Oncotarget 2016, 7, 74031–74042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Ardizzone, A.; Scuderi, S.A.; Giuffrida, D.; Colarossi, C.; Puglisi, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. Role of Fibroblast Growth Factors Receptors (FGFRs) in Brain Tumors, Focus on Astrocytoma and Glioblastoma. Cancers 2020, 12, 3825. [Google Scholar] [CrossRef] [PubMed]
- Girardi, F.; Matz, M.; Stiller, C.; You, H.; Marcos Gragera, R.; Valkov, M.Y.; Bulliard, J.L.; De, P.; Morrison, D.; Wanner, M.; et al. Global survival trends for brain tumors, by histology: Analysis of individual records for 556,237 adults diagnosed in 59 countries during 2000-2014 (CONCORD-3). Neuro Oncol. 2022, 67, 877. [Google Scholar] [CrossRef] [PubMed]
- Daubon, T.; Hemadou, A.; Romero Garmendia, I.; Saleh, M. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front Immunol. 2020, 11, 585616. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Chen, X.; Wei, P.; Lin, Y.; Wu, Z.; Lin, Z.; Kang, D.; Ding, C. Single-cell RNA sequencing reveals intra-tumoral heterogeneity of glioblastoma and a pro-tumor subset of tumor-associated macrophages characterized by EZH2 overexpression. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166534. [Google Scholar] [CrossRef]
- Fu, L.; Chen, S.; He, G.; Chen, Y.; Liu, B. Targeting Extracellular Signal-Regulated Protein Kinase 1/2 (ERK1/2) in Cancer: An Update on Pharmacological Small-Molecule Inhibitors. J. Med. Chem. 2022, 65, 13561–13573. [Google Scholar] [CrossRef]
- Sun, T.; Liu, Z.; Yang, Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer 2020, 19, 146. [Google Scholar] [CrossRef]
- Gharwan, H.; Groninger, H. Kinase inhibitors and monoclonal antibodies in oncology: Clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef]
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Nakajima, K. Role of the Immune System in the Development of the Central Nervous System. Front Neurosci. 2019, 13, 916. [Google Scholar] [CrossRef] [Green Version]
- Feng, E.; Liang, T.; Wang, X.; Du, J.; Tang, K.; Wang, X.; Wang, F.; You, G. Correlation of alteration of HLA-F expression and clinical characterization in 593 brain glioma samples. J. Neuroinflammation 2019, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Burster, T.; Gartner, F.; Bulach, C.; Zhanapiya, A.; Gihring, A.; Knippschild, U. Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells. Pharmaceuticals 2021, 14, 236. [Google Scholar] [CrossRef] [PubMed]
- Chung, R.; Whaley, J.; Kley, N.; Anderson, K.; Louis, D.; Menon, A.; Hettlich, C.; Freiman, R.; Hedley-Whyte, E.T.; Martuza, R.; et al. TP53 gene mutations and 17p deletions in human astrocytomas. Genes Chromosomes Cancer 1991, 3, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.; Depetris, D.; Iovanna, J.L.; Mattei, M.G.; Pebusque, M.J. Assignment of the tumor protein p53 induced nuclear protein 2 (TP53INP2) gene to human chromosome band 20q11.2 by in situ hybridization. Cytogenet. Genome Res. 2005, 108, 362. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase-Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Ko, Y.T. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch. Pharm. Res. 2020, 43, 385–394. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Janne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol Res. 2020, 152, 104609. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Nakada, M.; Kita, D.; Teng, L.; Pyko, I.V.; Watanabe, T.; Hayashi, Y.; Hamada, J.I. Receptor Tyrosine Kinases: Principles and Functions in Glioma Invasion. Adv. Exp. Med. Biol. 2020, 1202, 151–178. [Google Scholar] [CrossRef]
- Mall, R.; Bynigeri, R.R.; Karki, R.; Malireddi, R.K.S.; Sharma, B.R.; Kanneganti, T.D. Pancancer transcriptomic profiling identifies key PANoptosis markers as therapeutic targets for oncology. NAR Cancer 2022, 4, zcac033. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, R.; Sato, M.; Morimoto, Y.; Ohara, K.; Kosugi, K.; Oishi, Y.; Kuranari, Y.; Murase, M.; Yoshida, K.; Toda, M. Quantitative assessment and clinical relevance of VEGFRs-positive tumor cells in refractory brain tumors. Exp. Mol. Pathol. 2020, 114, 104408. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.W.; Liu, D.K.; Zhang, Q.W.; Xu, Y.G.; Shi, L. VEGFR-2 inhibitors and the therapeutic applications thereof: A patent review (2012–2016). Expert. Opin. Ther. Pat. 2017, 27, 987–1004. [Google Scholar] [CrossRef]
- Czabanka, M.; Bruenner, J.; Parmaksiz, G.; Broggini, T.; Topalovic, M.; Bayerl, S.H.; Auf, G.; Kremenetskaia, I.; Nieminen, M.; Jabouille, A.; et al. Combined temozolomide and sunitinib treatment leads to better tumour control but increased vascular resistance in O6-methylguanine methyltransferase-methylated gliomas. Eur. J. Cancer 2013, 49, 2243–2252. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Huang, Y.K.; Su, Y.F.; Lieu, A.S.; Loh, J.K.; Li, C.Y.; Wu, C.H.; Kuo, K.L.; Lin, C.L. MiR-1271 regulates glioblastoma cell proliferation and invasion by directly targeting the CAMKK2 gene. Neurosci. Lett. 2020, 737, 135289. [Google Scholar] [CrossRef]
- Hertler, C.; Seystahl, K.; Le Rhun, E.; Wirsching, H.G.; Roth, P.; Weller, M.; Gramatzki, D. Improved seizure control in patients with recurrent glioblastoma treated with bevacizumab. Neuro Oncol. 2022, 24, 2001–2004. [Google Scholar] [CrossRef]
- de Groot, J.F.; Lamborn, K.R.; Chang, S.M.; Gilbert, M.R.; Cloughesy, T.F.; Aldape, K.; Yao, J.; Jackson, E.F.; Lieberman, F.; Robins, H.I.; et al. Phase II study of aflibercept in recurrent malignant glioma: A North American Brain Tumor Consortium study. J. Clin. Oncol. 2011, 29, 2689–2695. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Wang, H.; Chen, G.; Zhou, Y. Anti-VEGFR2 monoclonal antibody(MSB0254) inhibits angiogenesis and tumor growth by blocking the signaling pathway mediated by VEGFR2 in glioblastoma. Biochem. Biophys. Res. Commun. 2022, 604, 158–164. [Google Scholar] [CrossRef]
- Awada, G.; Ben Salama, L.; De Cremer, J.; Schwarze, J.K.; Fischbuch, L.; Seynaeve, L.; Du Four, S.; Vanbinst, A.M.; Michotte, A.; Everaert, H.; et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: A stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J. Immunother. Cancer 2020, 8, e001146. [Google Scholar] [CrossRef]
- Du Four, S.; Maenhout, S.K.; Benteyn, D.; De Keersmaecker, B.; Duerinck, J.; Thielemans, K.; Neyns, B.; Aerts, J.L. Disease progression in recurrent glioblastoma patients treated with the VEGFR inhibitor axitinib is associated with increased regulatory T cell numbers and T cell exhaustion. Cancer Immunol. Immunother. 2016, 65, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Algahtani, M.; Natarajan, U.; Alhazzani, K.; Alaseem, A.; Rathinavelu, A. Evaluation of anti-angiogenic agent F16 for targeting glioblastoma xenograft tumors. Cancer Genet. 2022, 264–265, 71–89. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, Y.; Li, L.; Gao, C.; Liu, D.; Li, H.; Zhao, Z.; Zhao, B. Pyrotinib-based treatments in HER2-positive breast cancer patients with brain metastases. Ann. Med. 2022, 54, 3085–3095. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C.; Schiff, R. HER2: Biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Pestalozzi, B.C.; Zahrieh, D.; Price, K.N.; Holmberg, S.B.; Lindtner, J.; Collins, J.; Crivellari, D.; Fey, M.F.; Murray, E.; Pagani, O.; et al. Identifying breast cancer patients at risk for Central Nervous System (CNS) metastases in trials of the International Breast Cancer Study Group (IBCSG). Ann. Oncol. 2006, 17, 935–944. [Google Scholar] [CrossRef]
- Lin, N.U.; Winer, E.P. Brain metastases: The HER2 paradigm. Clin. Cancer Res. 2007, 13, 1648–1655. [Google Scholar] [CrossRef]
- Kaplan, H.G.; Malmgren, J.A.; Guo, B.; Atwood, M.K. Trastuzumab therapy duration in HER2-positive de novo metastatic breast cancer: 1999-2018. Breast Cancer Res. Treat 2022, 195, 171–180. [Google Scholar] [CrossRef]
- Oberkampf, F.; Gutierrez, M.; Trabelsi Grati, O.; Le Rhun, E.; Tredan, O.; Turbiez, I.; Kadi, A.; Dubot, C.; Taillibert, S.; Vacher, S.; et al. Phase II study of intrathecal administration of trastuzumab in patients with HER2-positive breast cancer with leptomeningeal metastasis. Neuro Oncol. 2022, 46, 890. [Google Scholar] [CrossRef]
- Wildiers, H.; Meyskens, T.; Marreaud, S.; Lago, L.D.; Vuylsteke, P.; Curigliano, G.; Waters, S.; Brouwers, B.; Meulemans, B.; Sousa, B.; et al. Long term outcome data from the EORTC 75111-10114 ETF/BCG randomized phase II study: Pertuzumab and trastuzumab with or without metronomic chemotherapy for older patients with HER2-positive metastatic breast cancer, followed by T-DM1 after progression. Breast 2022, 64, 100–111. [Google Scholar] [CrossRef]
- Petrelli, F.; Ghidini, M.; Lonati, V.; Tomasello, G.; Borgonovo, K.; Ghilardi, M.; Cabiddu, M.; Barni, S. The efficacy of lapatinib and capecitabine in HER-2 positive breast cancer with brain metastases: A systematic review and pooled analysis. Eur. J. Cancer 2017, 84, 141–148. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, X.; Chen, L.; Yin, Z.; Zuo, Z. Discovery and evaluation of cytisine N-isoflavones as novel EGFR/HER2 dual inhibitors. Bioorg. Chem. 2022, 127, 105868. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, A.R.; Greenland, E.L.; Pixley, F.J. Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers 2017, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Liaw, K.; Reddy, R.; Sharma, A.; Li, J.; Chang, M.; Sharma, R.; Salazar, S.; Kannan, S.; Kannan, R.M. Targeted systemic dendrimer delivery of CSF-1R inhibitor to tumor-associated macrophages improves outcomes in orthotopic glioblastoma. Bioeng Transl. Med. 2021, 6, e10205. [Google Scholar] [CrossRef]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Lin, C.-C.; Gil-Martin, M.; Bauer, T.M.; Naing, A.; Lim, D.W.-T.; Sarantopoulos, J.; Geva, R.; Ando, Y.; Fan, L.; Choudhury, S. Abstract CT171: Phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 2020, 80, CT171. [Google Scholar] [CrossRef]
- Almahariq, M.F.; Quinn, T.J.; Kesarwani, P.; Kant, S.; Miller, C.R.; Chinnaiyan, P. Inhibition of Colony-Stimulating Factor-1 Receptor Enhances the Efficacy of Radiotherapy and Reduces Immune Suppression in Glioblastoma. Vivo 2021, 35, 119–129. [Google Scholar] [CrossRef]
- Edwards, V.D.; Sweeney, D.T.; Ho, H.; Eide, C.A.; Rofelty, A.; Agarwal, A.; Liu, S.Q.; Danilov, A.V.; Lee, P.; Chantry, D.; et al. Targeting of colony-stimulating factor 1 receptor (CSF1R) in the CLL microenvironment yields antineoplastic activity in primary patient samples. Oncotarget 2018, 9, 24576–24589. [Google Scholar] [CrossRef] [Green Version]
- Akazawa, Y.; Kono, H.; Hara, M.; Furuya, S.; Nakata, Y.; Wakana, H.; Fukushima, H.; Sun, C.; Fujii, H.; Ichikawa, D. M-CSF Receptor Antagonists Inhibit the Initiation and Progression of Hepatocellular Carcinoma in Mice. Anticancer Res. 2019, 39, 4787–4794. [Google Scholar] [CrossRef]
- Blazquez, R.; Wlochowitz, D.; Wolff, A.; Seitz, S.; Wachter, A.; Perera-Bel, J.; Bleckmann, A.; Beissbarth, T.; Salinas, G.; Riemenschneider, M.J.; et al. PI3K: A master regulator of brain metastasis-promoting macrophages/microglia. Glia 2018, 66, 2438–2455. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-gamma and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control Release 2020, 321, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Knight, Z.A.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Hornschemeyer, J.; Kirschstein, T.; Reichart, G.; Sasse, C.; Venus, J.; Einsle, A.; Porath, K.; Linnebacher, M.; Kohling, R.; Lange, F. Studies on Biological and Molecular Effects of Small-Molecule Kinase Inhibitors on Human Glioblastoma Cells and Organotypic Brain Slices. Life 2022, 12, 1258. [Google Scholar] [CrossRef]
- de Gooijer, M.C.; Zhang, P.; Buil, L.C.M.; Citirikkaya, C.H.; Thota, N.; Beijnen, J.H.; van Tellingen, O. Buparlisib is a brain penetrable pan-PI3K inhibitor. Sci. Rep. 2018, 8, 10784. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Montagut, C.; Settleman, J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009, 283, 125–134. [Google Scholar] [CrossRef]
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X.; et al. FDA Approval Summary: Dabrafenib and Trametinib for the Treatment of Metastatic Non-Small Cell Lung Cancers Harboring BRAF V600E Mutations. Oncologist 2018, 23, 740–745. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Swanner, J.; Otani, Y.; Nair, M.; Park, F.; Banasavadi-Siddegowda, Y.; Liu, J.; Jaime-Ramirez, A.C.; Hong, B.; Geng, F.; et al. Oncolytic HSV therapy increases trametinib access to brain tumors and sensitizes them in vivo. Neuro Oncol. 2019, 21, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Huang, Y.; Semtner, M.; Zhao, K.; Tan, Z.; Dzaye, O.; Kettenmann, H.; Shu, K.; Lei, T. Down-regulation of Aquaporin-1 mediates a microglial phenotype switch affecting glioma growth. Exp. Cell Res. 2020, 396, 112323. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Bae, K.; Baek, A.R.; Kwon, E.B.; Kim, Y.H.; Nam, S.W.; Lee, G.H.; Chang, Y. Glioblastoma-Derived Exosomes as Nanopharmaceutics for Improved Glioma Treatment. Pharmaceutics 2022, 14, 1002. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Dey, M.; Chang, A.; Lesniak, M.S. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front Immunol. 2013, 4, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comba, A.; Dunn, P.J.; Argento, A.E.; Kadiyala, P.; Ventosa, M.; Patel, P.; Zamler, D.B.; Nunez, F.J.; Zhao, L.; Castro, M.G.; et al. Fyn tyrosine kinase, a downstream target of receptor tyrosine kinases, modulates antiglioma immune responses. Neuro Oncol. 2020, 22, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanachetty, S.; Cruz-Cruz, J.; Hoffmeyer, E.; Cole, A.P.; Mitra, S.S. New Insights into the Multifaceted Role of Myeloid-Derived Suppressor Cells (MDSCs) in High-Grade Gliomas: From Metabolic Reprograming, Immunosuppression, and Therapeutic Resistance to Current Strategies for Targeting MDSCs. Cells 2021, 10, 893. [Google Scholar] [CrossRef] [PubMed]
- Filippone, A.; Lanza, M.; Mannino, D.; Raciti, G.; Colarossi, C.; Sciacca, D.; Cuzzocrea, S.; Paterniti, I. PD1/PD-L1 immune checkpoint as a potential target for preventing brain tumor progression. Cancer Immunol. Immunother. 2022, 71, 2067–2075. [Google Scholar] [CrossRef]
- DiDomenico, J.; Lamano, J.B.; Oyon, D.; Li, Y.; Veliceasa, D.; Kaur, G.; Ampie, L.; Choy, W.; Lamano, J.B.; Bloch, O. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 2018, 7, e1448329. [Google Scholar] [CrossRef] [Green Version]
- Wintterle, S.; Schreiner, B.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Meyermann, R.; Weller, M.; Wiendl, H. Expression of the B7-related molecule B7-H1 by glioma cells: A potential mechanism of immune paralysis. Cancer Res. 2003, 63, 7462–7467. [Google Scholar]
- Lee, J.W.; Park, B.C.; Jang, N.Y.; Lee, S.; Cho, Y.K.; Sharma, P.; Byun, S.W.; Jeon, K.; Jeon, Y.H.; Park, U.; et al. Inducing Ectopic T Cell Clusters Using Stromal Vascular Fraction Spheroid-Based Immunotherapy to Enhance Anti-Tumor Immunity. Adv. Sci. 2022, 9, e2203842. [Google Scholar] [CrossRef]
- Ozao-Choy, J.; Ma, G.; Kao, J.; Wang, G.X.; Meseck, M.; Sung, M.; Schwartz, M.; Divino, C.M.; Pan, P.Y.; Chen, S.H. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009, 69, 2514–2522. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Rayman, P.; Huang, P.; Grabowski, M.; Hambardzumyan, D.; Finke, J.H.; Vogelbaum, M.A. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J. Neurooncol. 2015, 122, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, S.; Sasaki, M.; Scharer, C.D.; Kissick, H.T.; Patterson, D.G.; Magliocca, K.R.; Seykora, J.T.; Sapkota, B.; Gutman, D.A.; Cooper, L.A. Phosphoinositide 3-kinase signaling can modulate MHC class I and II expression. Mol. Cancer Res. 2019, 17, 2395–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, M.; Schulz, D.; Piendl, G.; Brockhoff, G.; Eichberger, J.; Menevse, A.-N.; Beckhove, P.; Hautmann, M.; Reichert, T.E.; Ettl, T. Buparlisib modulates PD-L1 expression in head and neck squamous cell carcinoma cell lines. Exp. Cell Res. 2020, 396, 112259. [Google Scholar] [CrossRef]
- Rausch, M.; Tchaicha, J.; Tibbitts, T.; Henau, O.D.; Sharma, S.; Pink, M.; Gladstone, J.; Proctor, J.; Douglas, M.; Stern, H. Abstract B032: The PI3K-γ inhibitor, IPI-549, increases antitumor immunity by targeting tumor-associated myeloid cells and remodeling the immune-suppressive tumor microenvironment. Cancer Immunol. Res. 2016, 4, B032. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, L.; Liu, Q.; Hou, L.; Huang, L. Inhibiting PI3 kinase-γ in both myeloid and plasma cells remodels the suppressive tumor microenvironment in desmoplastic tumors. J. Control. Release 2019, 309, 173–180. [Google Scholar] [CrossRef]
- Miyazaki, T.; Ishikawa, E.; Matsuda, M.; Sugii, N.; Kohzuki, H.; Akutsu, H.; Sakamoto, N.; Takano, S.; Matsumura, A. Infiltration of CD163-positive macrophages in glioma tissues after treatment with anti-PD-L1 antibody and role of PI3Kgamma inhibitor as a combination therapy with anti-PD-L1 antibody in in vivo model using temozolomide-resistant murine glioma-initiating cells. Brain Tumor Pathol. 2020, 37, 41–49. [Google Scholar] [CrossRef]
- Nasirzadeh, M.; Rasmi, Y.; Rahbarghazi, R.; Kheradmand, F.; Karimipour, M.; Aramwit, P.; Astinfeshan, M.; Gholinejad, Z.; Daeihasani, B.; Saboory, E.; et al. Crocetin promotes angiogenesis in human endothelial cells through PI3K-Akt-eNOS signaling pathway. EXCLI J. 2019, 18, 936–949. [Google Scholar] [CrossRef]
- Takada, S.; Hashishita, H.; Nagamori, S.; Endo, M. Axitinib-Induced Hypothyroidism as a Predictor of Long-Term Survival in Patients with Metastatic Renal Cell Carcinoma. Urol. Int. 2019, 102, 435–440. [Google Scholar] [CrossRef]
- Fu, Y.; Saxu, R.; Ridwan, K.A.; Yao, J.; Chen, X.; Xu, X.; Zheng, W.; Yu, P.; Teng, Y. Losartan Alleviates the Side Effects and Maintains the Anticancer Activity of Axitinib. Molecules 2022, 27, 2764. [Google Scholar] [CrossRef] [PubMed]
- Aw, D.C.; Tan, E.H.; Chin, T.M.; Lim, H.L.; Lee, H.Y.; Soo, R.A. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac. J. Clin. Oncol. 2018, 14, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernov, A.N.; Alaverdian, D.A.; Galimova, E.S.; Renieri, A.; Frullanti, E.; Meloni, I.; Shamova, O.V. The phenomenon of multidrug resistance in glioblastomas. Hematol. Oncol. Stem. Cell Ther. 2021, 12, 1004. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippone, A.; Mannino, D.; Casili, G.; Lanza, M.; Paterniti, I.; Cuzzocrea, S.; Capra, A.P.; Colarossi, L.; Giuffrida, D.; Lombardo, S.P.; et al. Protein Kinase Inhibitors as a New Target for Immune System Modulation and Brain Cancer Management. Int. J. Mol. Sci. 2022, 23, 15693. https://doi.org/10.3390/ijms232415693
Filippone A, Mannino D, Casili G, Lanza M, Paterniti I, Cuzzocrea S, Capra AP, Colarossi L, Giuffrida D, Lombardo SP, et al. Protein Kinase Inhibitors as a New Target for Immune System Modulation and Brain Cancer Management. International Journal of Molecular Sciences. 2022; 23(24):15693. https://doi.org/10.3390/ijms232415693
Chicago/Turabian StyleFilippone, Alessia, Deborah Mannino, Giovanna Casili, Marika Lanza, Irene Paterniti, Salvatore Cuzzocrea, Anna Paola Capra, Lorenzo Colarossi, Dario Giuffrida, Sofia Paola Lombardo, and et al. 2022. "Protein Kinase Inhibitors as a New Target for Immune System Modulation and Brain Cancer Management" International Journal of Molecular Sciences 23, no. 24: 15693. https://doi.org/10.3390/ijms232415693