1. Introduction
The complex world of fluorescent proteins (FP) is an intriguing and fascinating field of chemical research.
FPs are small and intrinsically fluorescent systems in which the chromophore forms autocatalytically in a host-independent process. These are among the unique features of FPs that permit their employment in a wide range of applications in cellular and molecular biology and biotechnology, along with the fact that their properties can be tuned [
1].
The growing interest in this class of proteins was first triggered by the discovery of the Green Fluorescent Protein in the jellyfish
Aequorea Victoria, which initiated the so-called GFP revolution [
2]. An eleven-stranded
-barrel structure and chromogenic XYG peptide are common in GFP-like proteins [
3]. Various color emitters are obtained by extending the chromophore conjugation length or tuning the 3D solvation shell, affecting both the cis–trans isomerization and the chromophore protonation state [
3,
4]. Further developments through mutagenesis can enhance the stability and expand the available spectral range, allowing the potential of GFP and related fluorescent proteins to be exploited [
5]. Despite a huge number of applications, FPs are complex systems, and their photochemistry is not yet completely understood.
Nowadays, thanks to modern spectroscopic techniques such as picosecond transient Raman Spectroscopy, time-resolved fluorescence [
6], femtosecond-stimulated Raman Spectroscopy [
7], and femtosecond UV spectroscopy [
8], it is possible to obtain major insights into photo-induced processes. The most studied system in the field of photo-reactivity is GFP, on account of the great interest in its huge number of applications and the complexity of its photochemistry. The chromophore
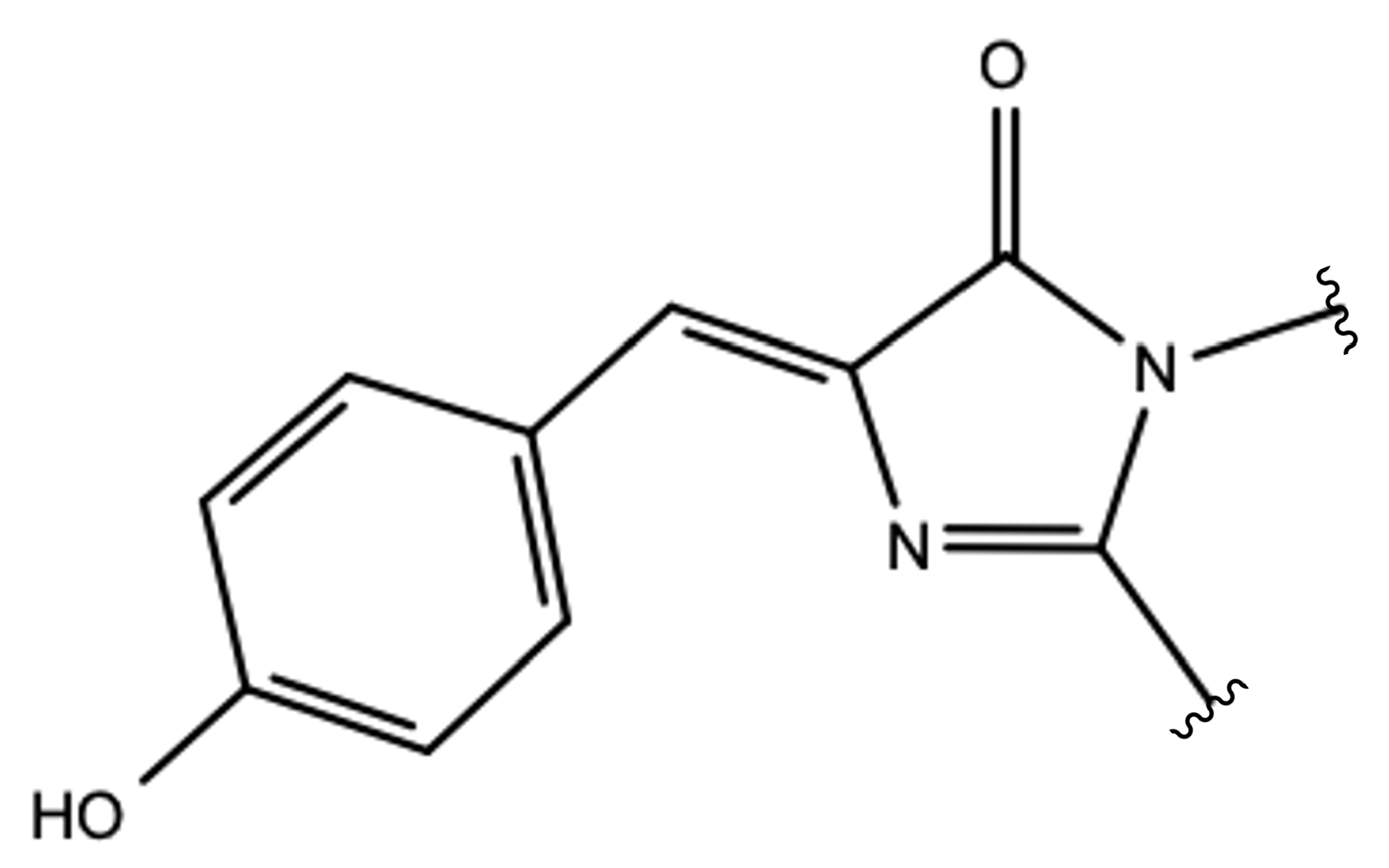
p-hydroxybenzylideneimidazolinone in its neutral form (HBDI; see
Figure 1) is responsible for an absorption band around 398 nm.
Upon electronic excitation, an excited-state proton transfer (ESPT) reaction takes place involving a hydrogen bond network composed of the chromophore, a crystallographic water molecule, and the residues Ser205 and Glu222 [
9]. It is the anionic form of the chromophore obtained from this reaction that is responsible for the bright fluorescence around 508 nm [
10]. Although many experimental and theoretical studies have been performed to clarify driving force of the ESPT along with its mechanism and kinetics, many open hypotheses exist and the available experimental data are insufficient to provide a complete picture of these phenomena. In this context, the theoretical-computational approach can be crucial to help in understanding the driving forces induced by the electronic excitation favoring the ESPT [
11,
12,
13,
14]. Despite this, thanks to their huge number of applications, studies on FPs are growing day by day, representing a great challenge from a scientific point of view.
The discovery in
Anthozoa of an entire family of fluorescent proteins and non-fluorescent chromoproteins distantly related to the GFP from
Aequorea Victoria has provided new tools that can be alternative or complement to the existing uses of GFP [
15]. Fluorescent proteins isolated from coral reef organisms in the
Anthozoa class can be grouped into five classes: red, yellow, cyan, green, and non-fluorescent chromoproteins. The general conclusion of the several studies on a variety of coral FPs is that the most important determinant of emission color is the physical extent of the conjugated portion of the chromophore. However, local environmental effects such as the position of the charged group near the chromophore, can lead to remarkable shifts in absorbance and emission maxima [
16,
17,
18]. Because of the high heterogeneity of the colors of such coral FPs, these systems are very interesting for a variety of in vivo techniques; moreover, they can be employed as models for experimental studies on the evolution of protein families [
15].
Within this family, the class of cyan fluorescent proteins (CFPs) possesses the same chromophore as the green fluorescent proteins [
16]. These proteins typically have an emission peak between 485–495 nm, although more blue-shifted variants can occasionally be found down to 477 nm, and exhibit absorption maxima from 430 and 460 nm. A cyan fluorescent protein exhibiting different behavior in comparison to other cyan fluorescent proteins, termed psamFP488, has been isolated from the genus
Psammocora of reef building corals [
19]. In particular, in this protein, unlike other cyan fluorescent proteins, a very blue-shifted absorption maximum is displayed around 404 nm, while the emission range is one typical of CFPs. Therefore, psamFP488 is characterized by an extended Stokes shift that is unexpected for CFPs. Moreover, the absorption maximum is very similar to that observed for wtGFP, making it reasonable to hypothesize similarities with the photo-chemical behavior of GFP.
In their work [
19], Kennis et al. performed experiments based on transient absorption spectroscopy and pump-dump-probe spectroscopy, and suggested that an ESPT reaction takes place upon electronic excitation that converts the neutral chromophore in its anionic form to the species responsible for the observed fluorescence peak.
The observed large Stokes shift should find its origin in this ESPT process. Furthermore, time-resolved experiments have revealed an ultrafast kinetics correlated to both the ESPT and the ground state proton back-transfer. In particular, experimental findings suggest that the ESPT reaction occurs in 170 fs, followed by a proton back-transfer that closes the photocycle and returns to the neutral chromophore in 110 fs.
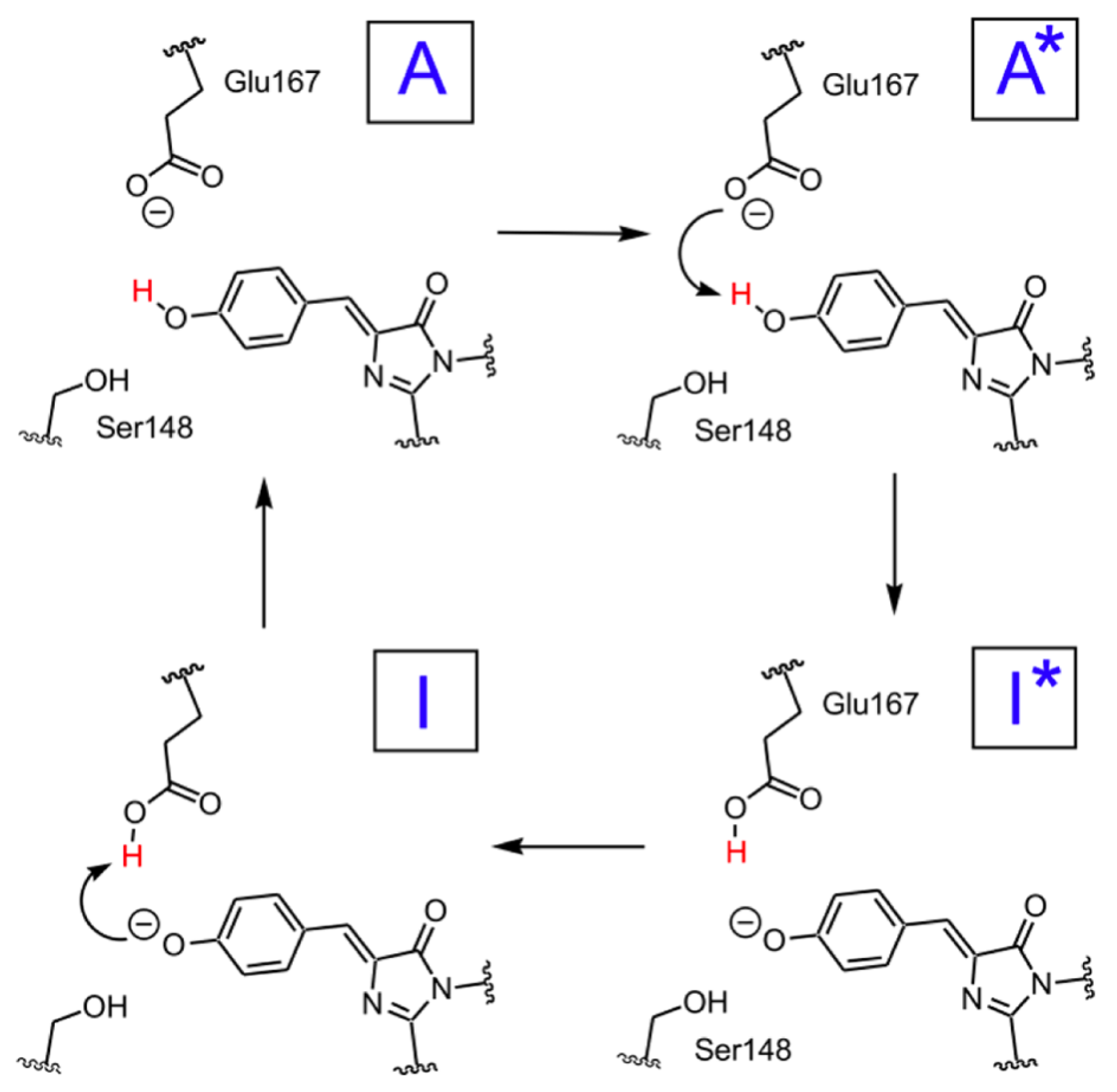
A scheme of the hypothesized photocycle for psamFP488 is shown in
Figure 2.
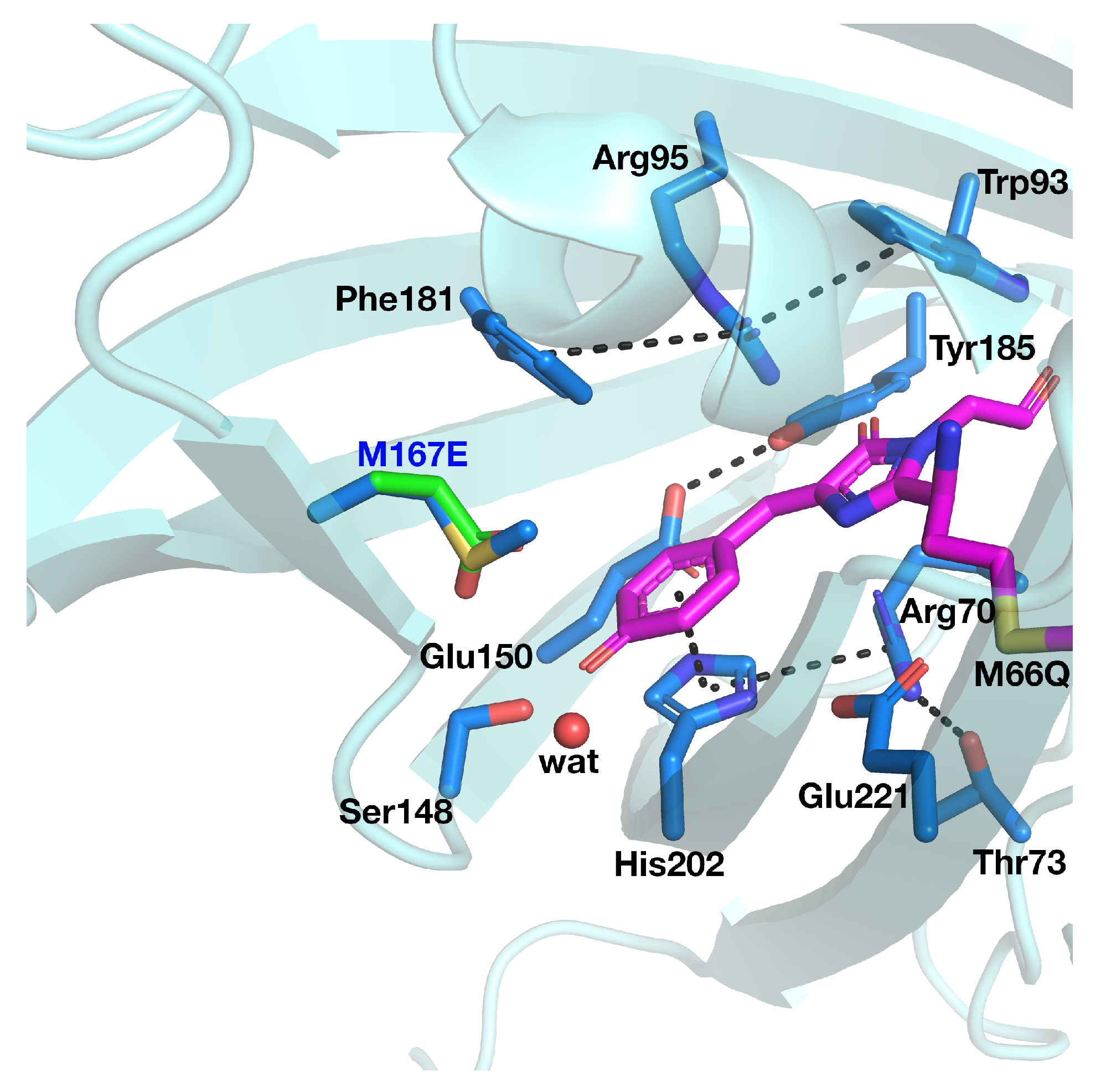
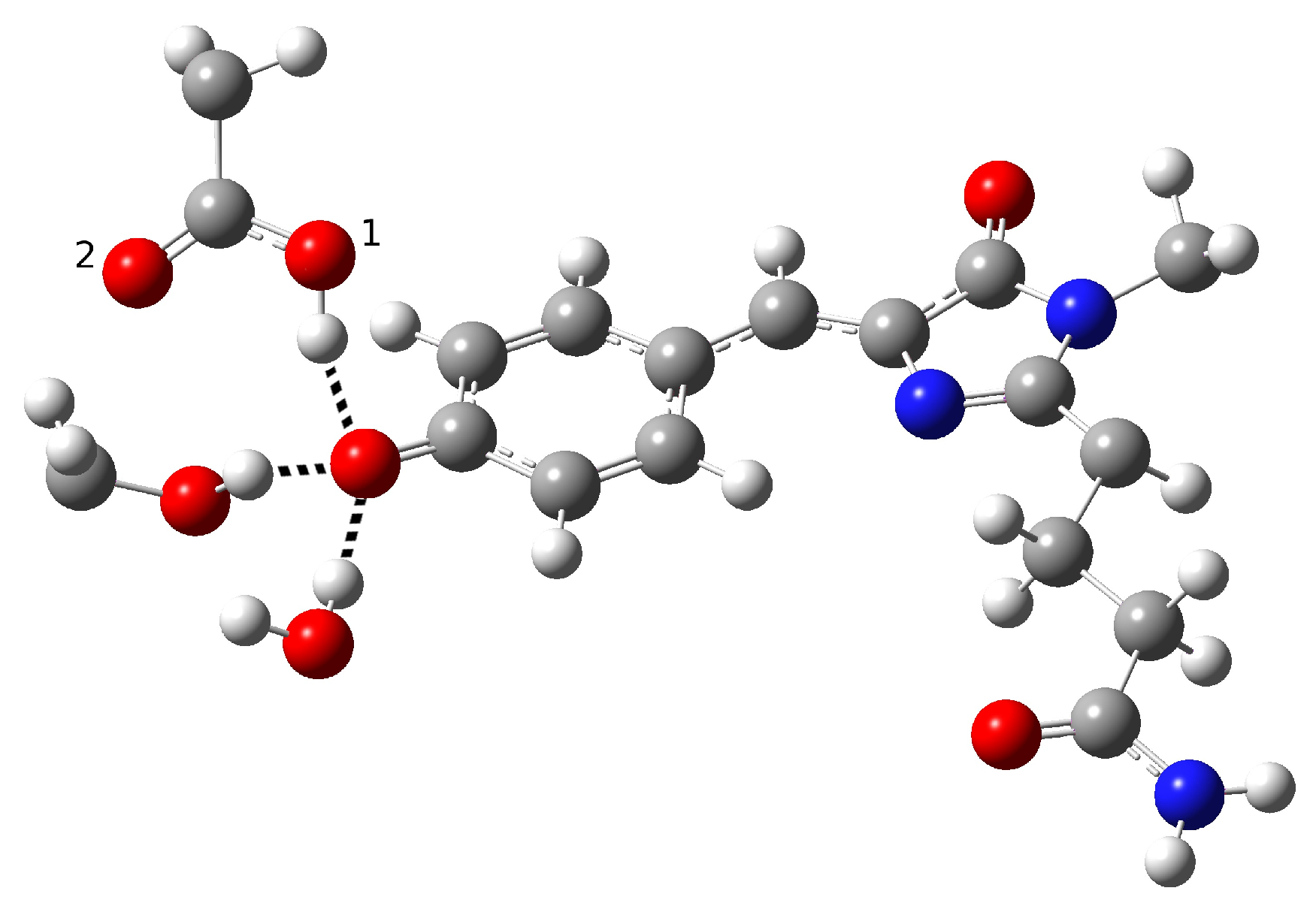
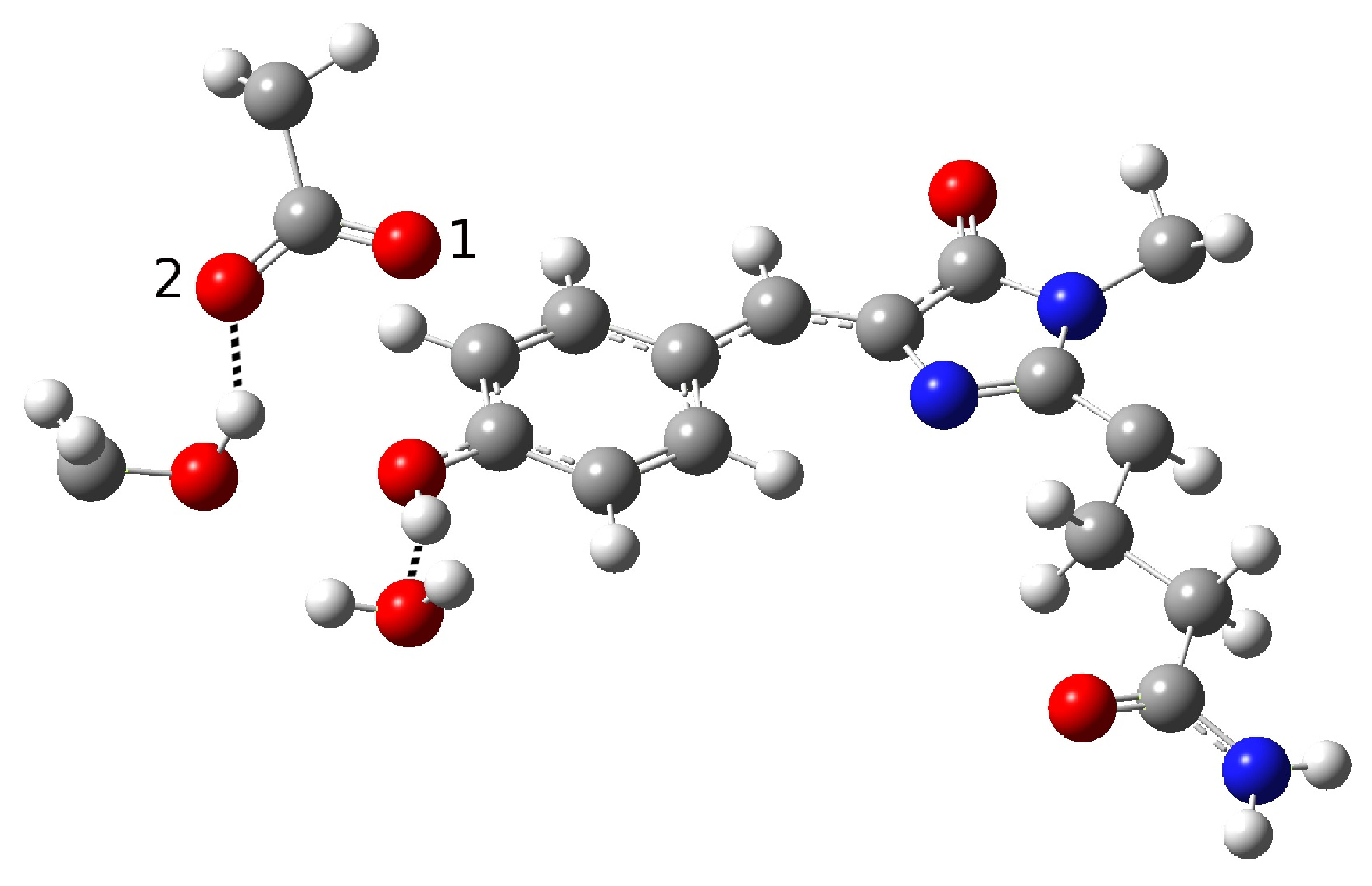
Although an ESPT has been hypothesized, the mechanism is not clear. It is possible that the ultrafast proton shuttling in the excited and ground states results from a favorable hydrogen-bonding geometry between the donor and acceptor. The hypothesis of a network surrounding the chromophore was proposed by Kennis et al. on the basis of the homologous zFP538-K66M shown in
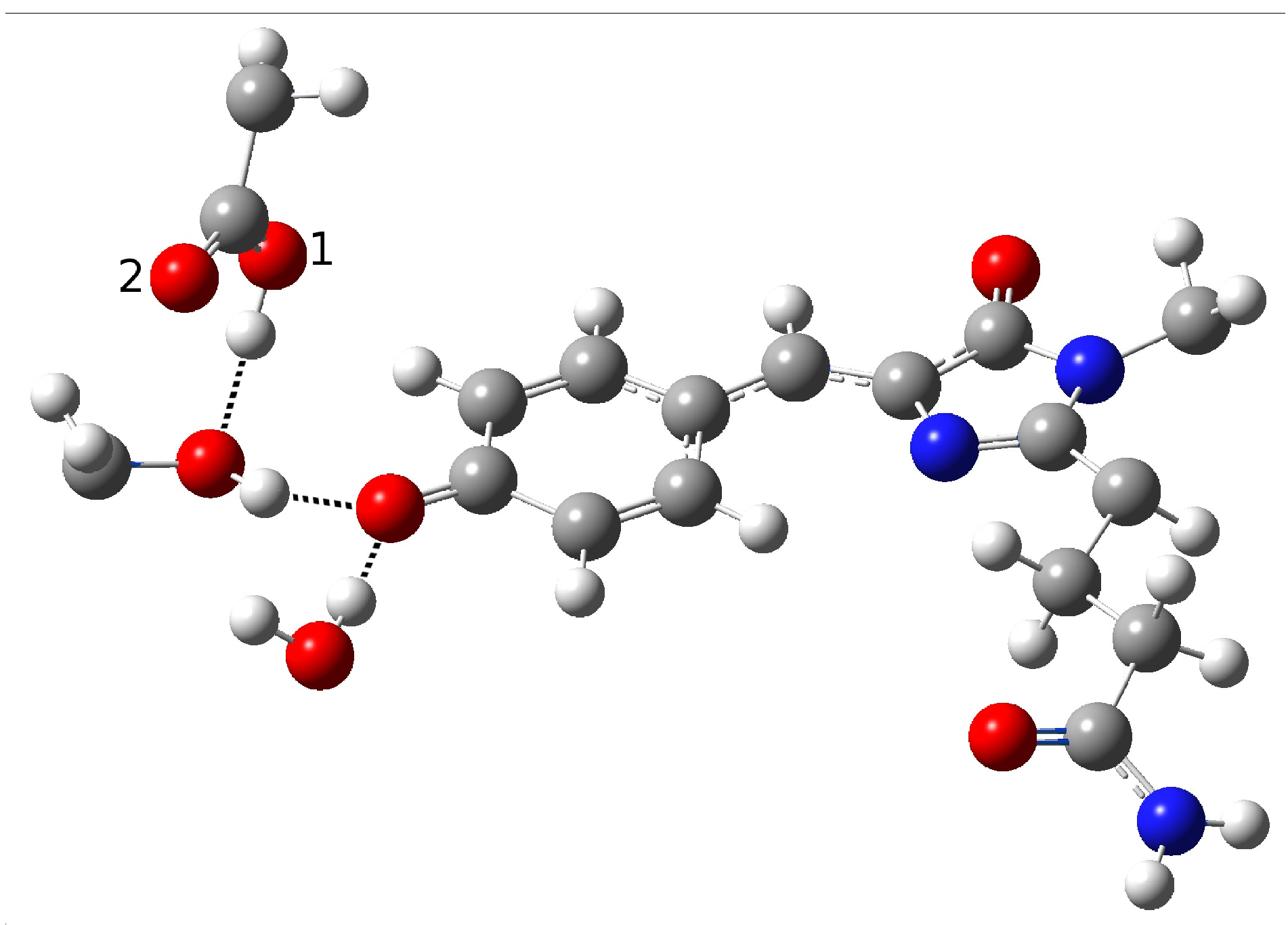
Figure 3 because of the lack of a crystallographic structure in psamFP488.
The main difference between psamFP488 and zFP538-K66M is represented by residue 167; in psamFP488, the methionine 167 is replaced by a glutammic acid. It is reasonable to suppose that Glu167, which seems to be very close to the chromophore, can act as proton acceptor. Previously, it has been noted that position 167 is crucial to tuning the cyan emission [
20].
However, the absence of a crystallographic structure in psamFP488, the intrinsic complexity in describing an excited state reaction, and the paucity of literature (both experimental and theoretical) on this system combine to make the current hypotheses and understanding of the reaction mechanism an open issue. An important contribution to provide insight on these problems, in particular with respect to obtaining a molecular picture of the experimental results, can be given by a theoretical-computational study.
In this study, a mixed quantum mechanics–molecular mechanics (QM/MM) approach is used to simulate the ESPT reaction by ab initio molecular dynamics in order to help in understanding the reaction mechanism starting from the hypotheses based on the experimental results.
In particular, because of the absence of a crystallographic structure in psamFP488, it is challenging to establish a reasonable starting configuration that can represent the first reactant of the photocycle; here, a computational approach can be helpful in hypothesizing plausible structural arrangements for the active site. In this way, different possibilities can be proposed and compared in order to obtain a reasonable starting point for the photocycle study.
Recently, it has been demonstrated at the theory level that a density functional theory (DFT) in its time-independent and time-dependent (TD) versions is able to provide a good description of theESPT reaction mechanism of GFP and the optical behavior of the chromophore in several environments [
12]. Encouraged by these results, we employ a similar protocol here to describe a novel and as yet not well understood system in psamFP488. In this case, the challenge is to characterize the photoactive species involved in the reaction starting from a reasonable hypothesis of the protein active site while building a solid model of the reactant despite the absence of a crystallographic protein structure. Our results suggest that, although an ultrafast ESPT could favor the hypothesis of a direct proton transfer from the chromophore to the final acceptor, other possible scenarios should be considered as well. Indeed, we suggest that a more complex and indirect mechanism could be both plausible and compatible with this CFP photo-induced reaction.
The rest of this paper is organized as follows: the main results are presented in the Results and Discussion section; the theoretical-computational strategy is described in the Materials and Methods section; finally, our main conclusions are provided in the Conclusions section.
4. Conclusions
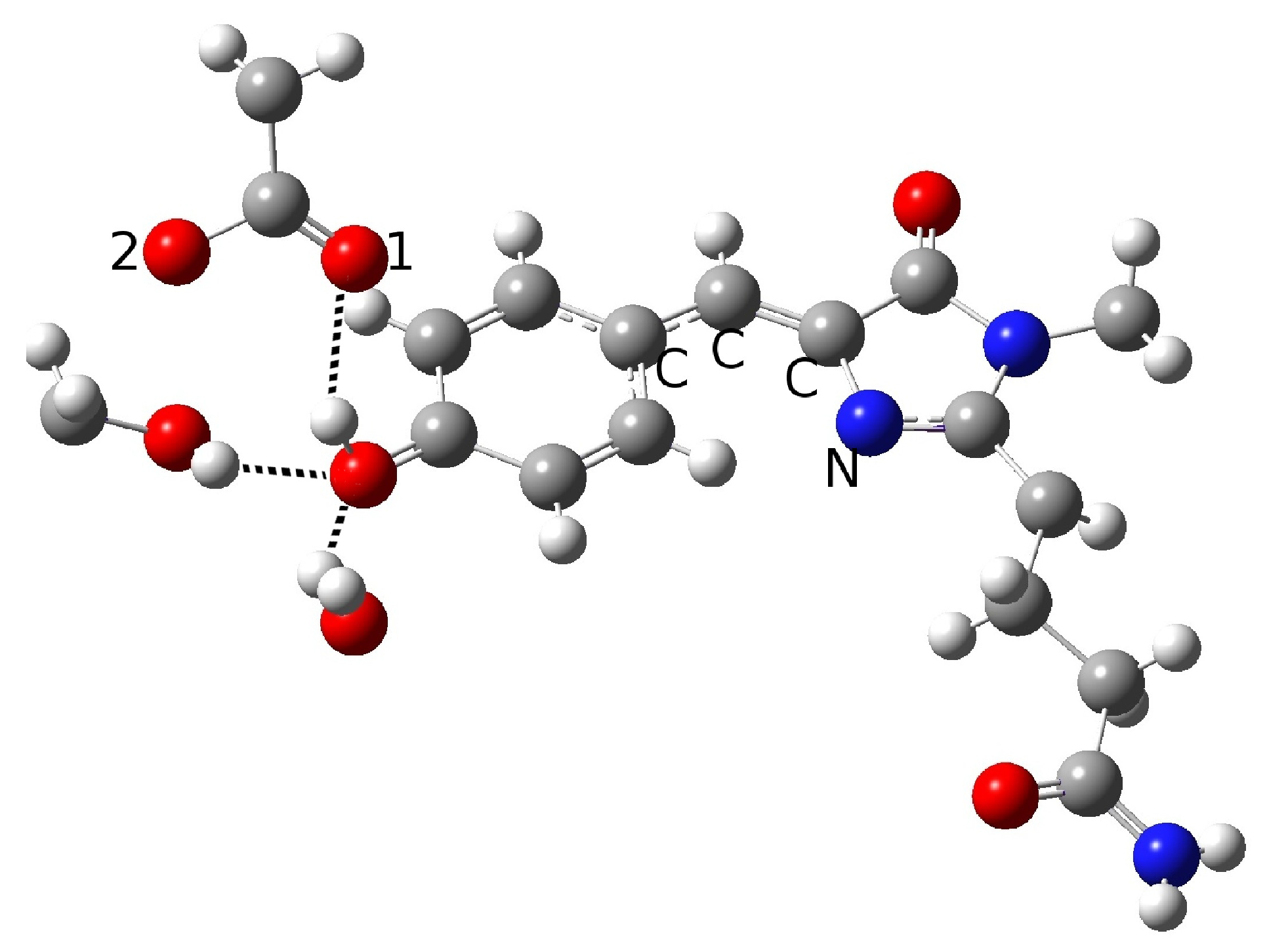
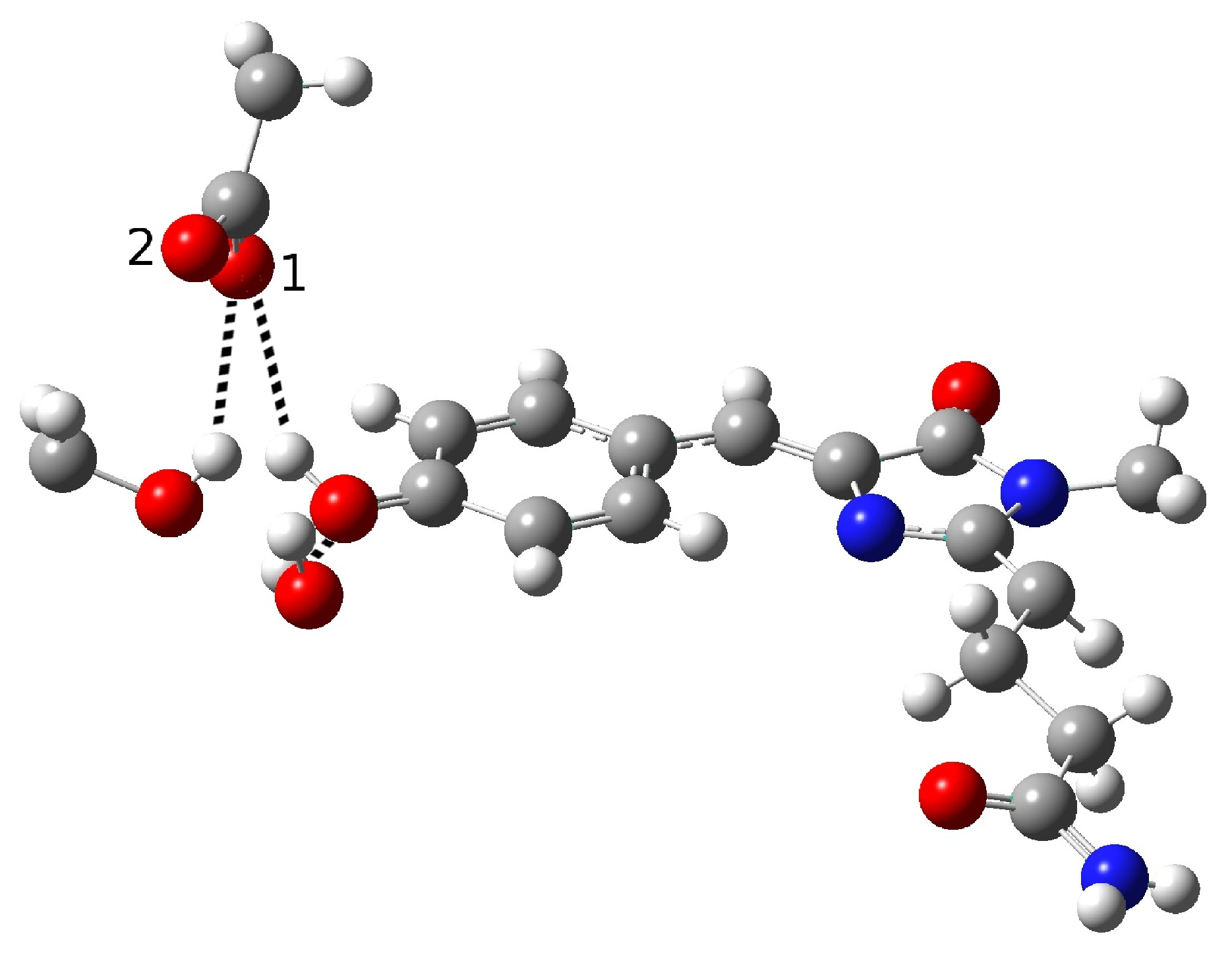
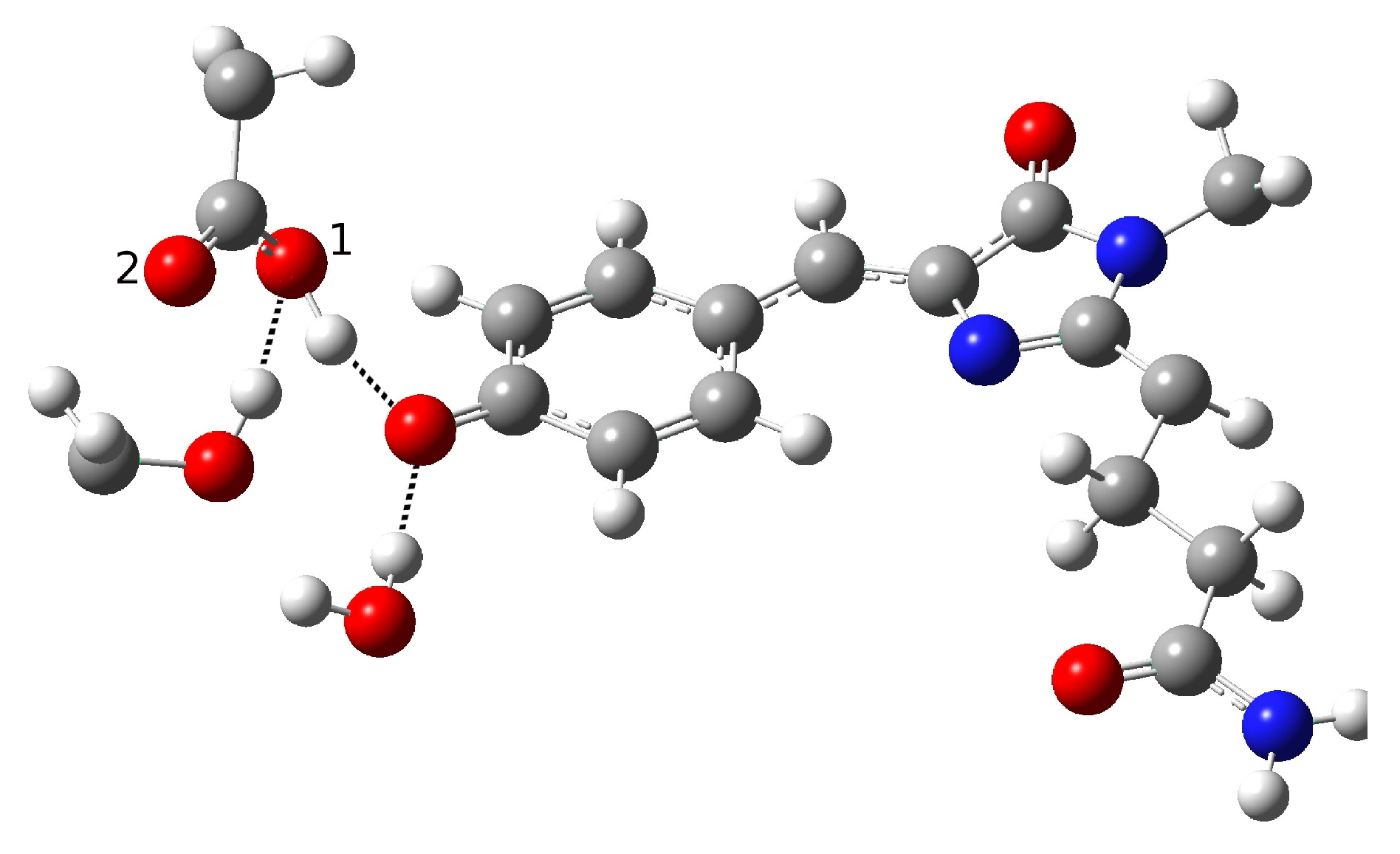
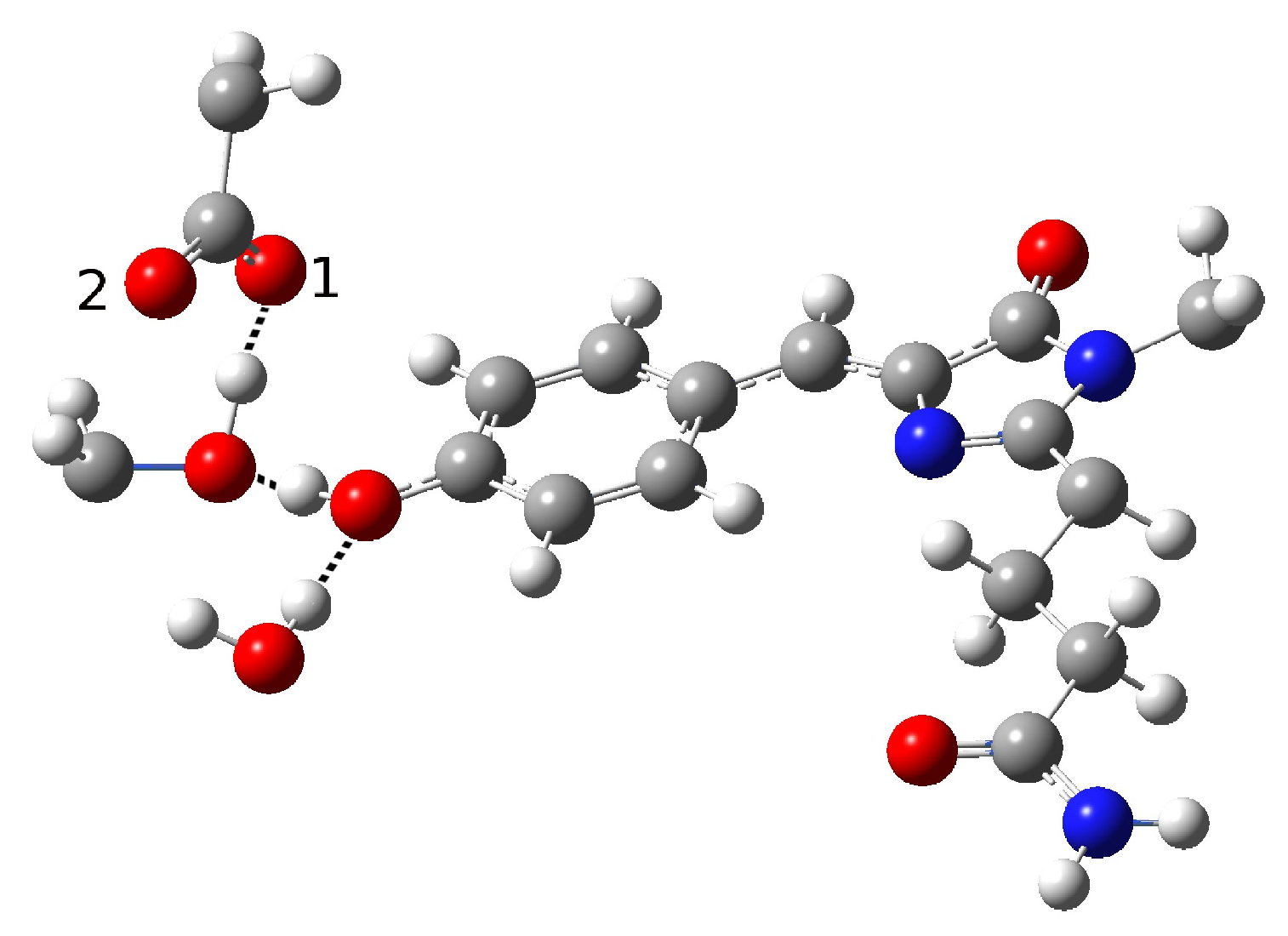
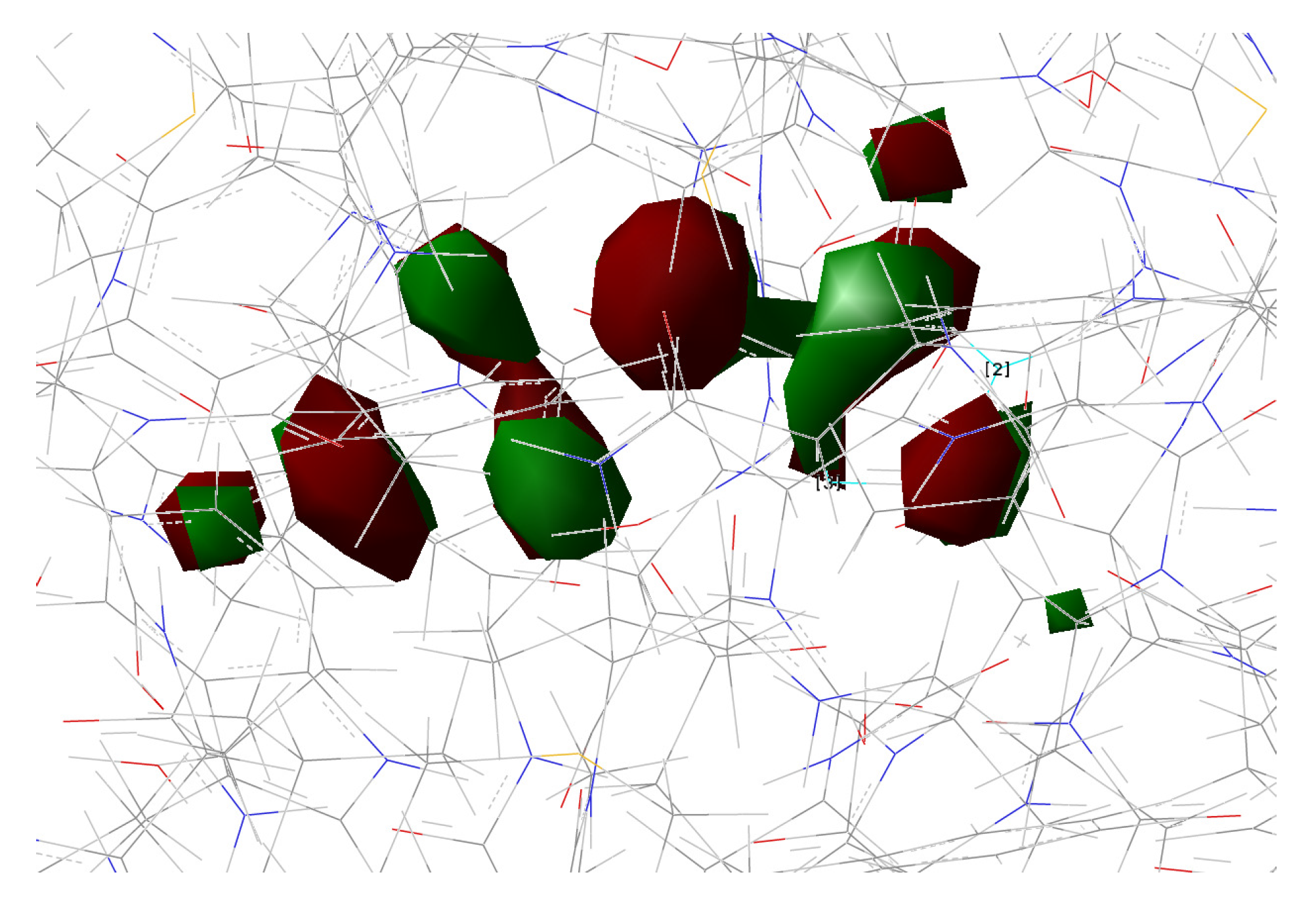
In the present work, we studied the photo-induced behavior of a new cyan fluorescent protein called psamFP488. In particular, we adopted a theoretical-computational approach in order to investigate possible alternative photocycle networks, starting from experimental hypotheses and then simulating the ESPT. We were not able to find a representative network with a neutral chromophore in the ground state when adopting the experimentally hypothesized arrangement implying a direct ESPT between the choromophore and Glu167. This evidence suggested that the experimental hypothesis of a direct Glu167–H(chromophore) interaction probably needed to be reviewed, and that a scenario involving a different hydrogen bonds network had to be considered.
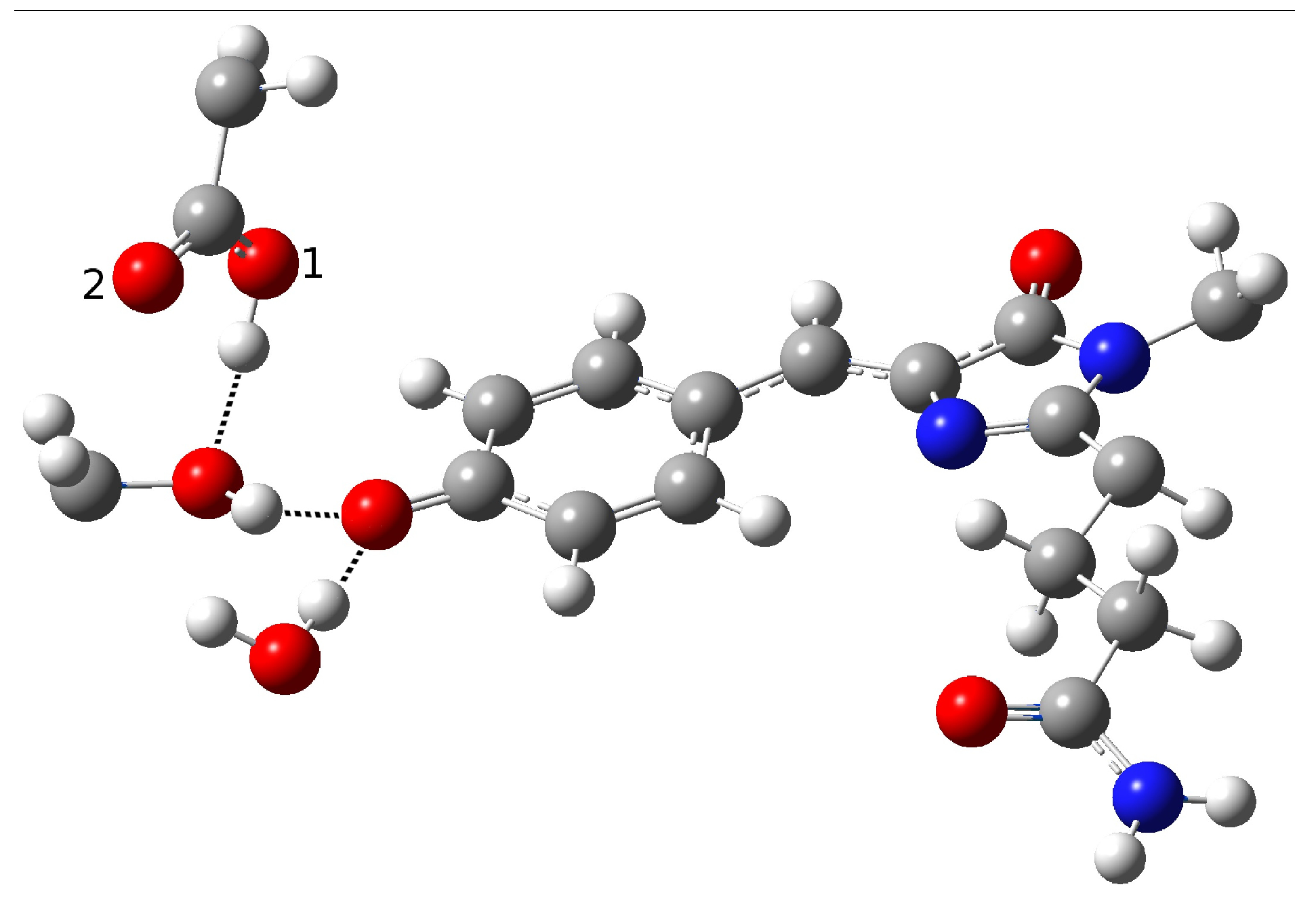
Therefore, we investigated possible alternative models to represent the first species taking part in the photocycle with a hydrogen bond pattern around the chromophore different from that experimentally hypothesized. The chosen arrangement (Network1) was able to properly reproduce the optical properties of the chromophore in its neutral form, corroborating the choice of this form as a possible starting arrangement for the ESPT reaction. On the basis of this newly proposed neutral form of the protein, it could be possible that the ESPT may not happen through direct interaction between donor and acceptor. Indeed, an excited state reaction mechanism involving a PT from the chromophore to Glu167 could be modulated by the Ser148 acting as a proton transfer relay station. Our study suggests that the experimental hypothesis of a direct Glu167–H(chromophore) interaction is less plausible considering the Franck–Condon region of the minimum energy structure defined by this HB network, although it is possible that, considering its dynamics at finite temperature, psamFP488 can visit a configurational space compatible with a stable neutral form and a direct ESPT between Glu167 and the chromophore.
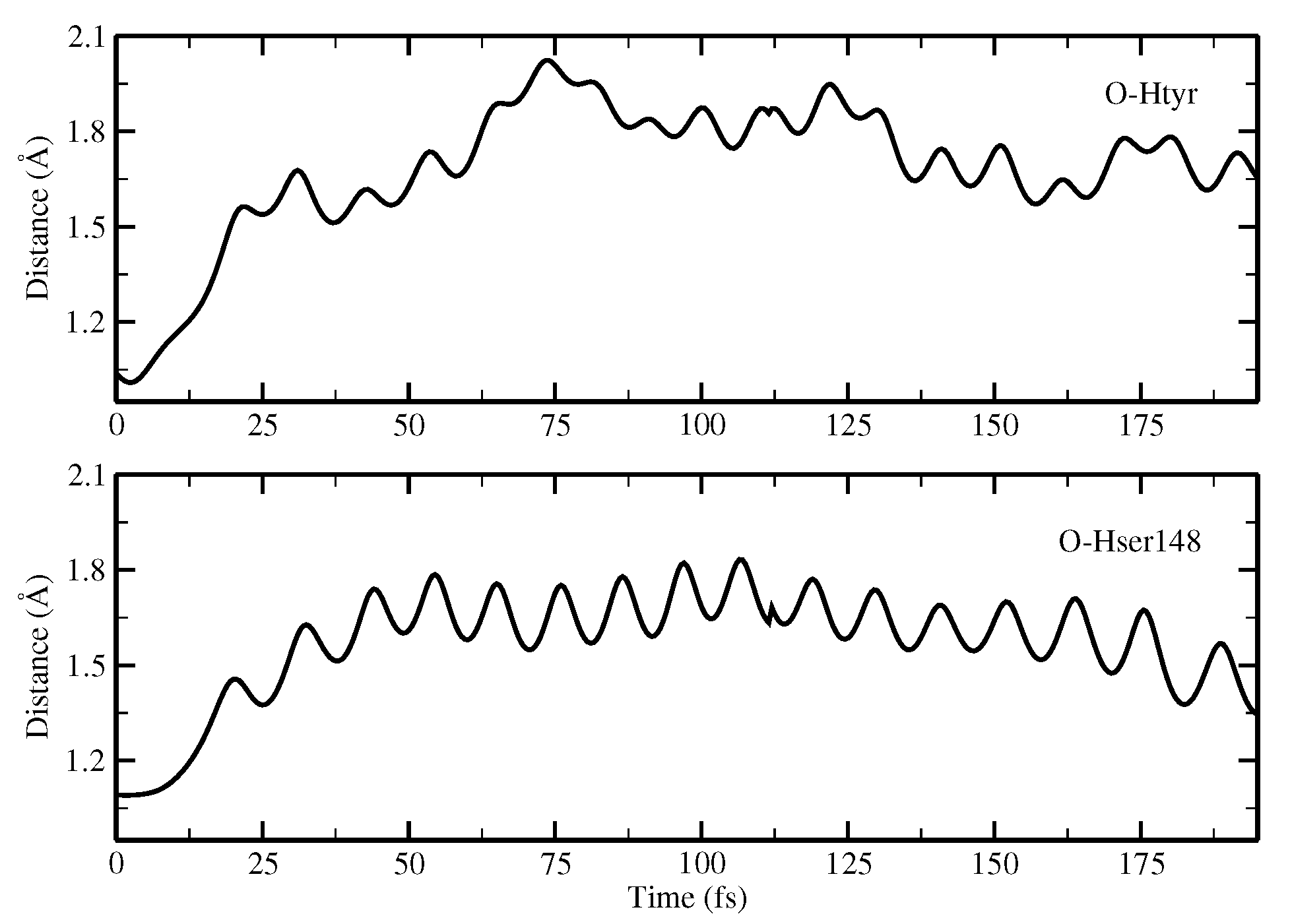
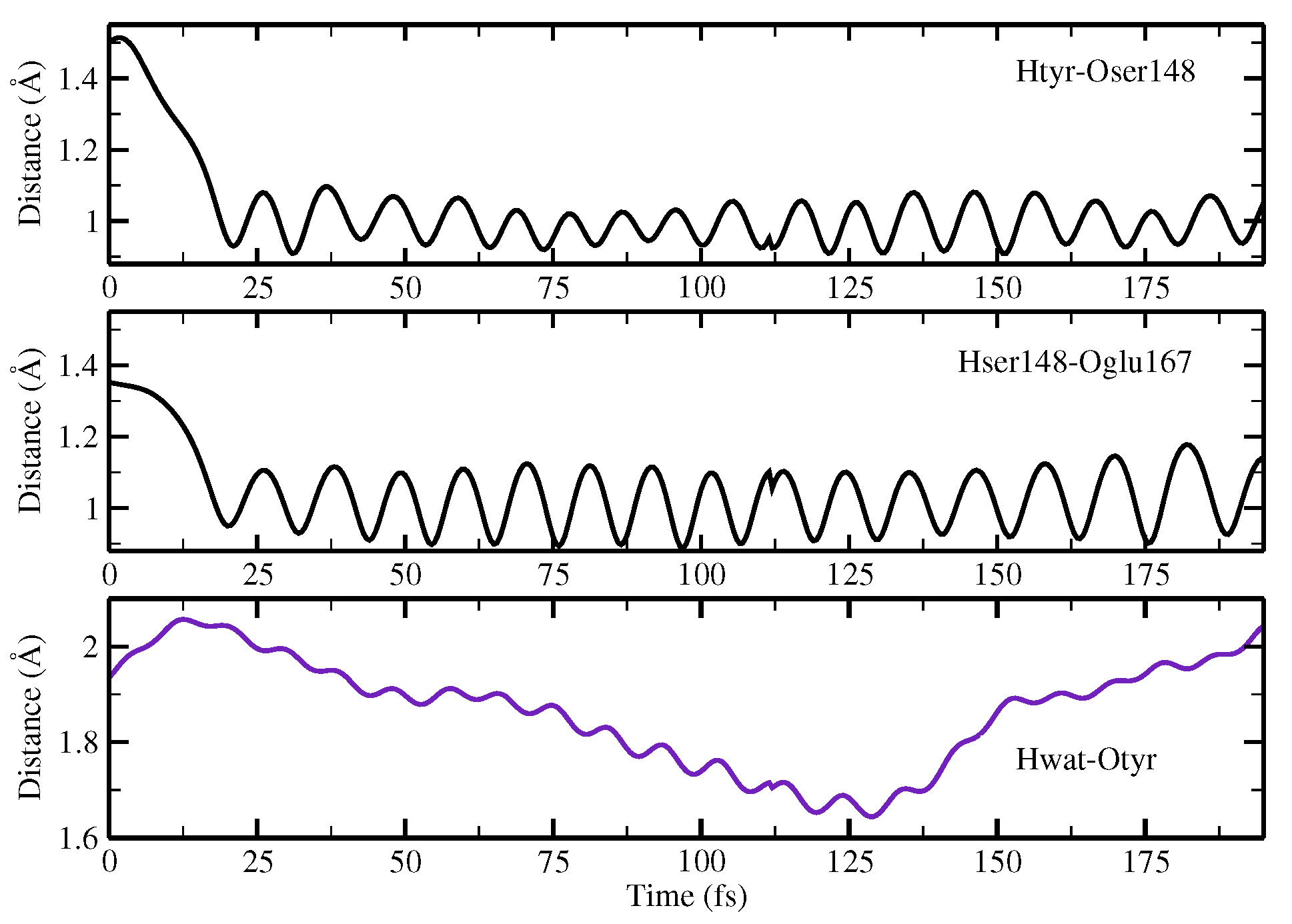
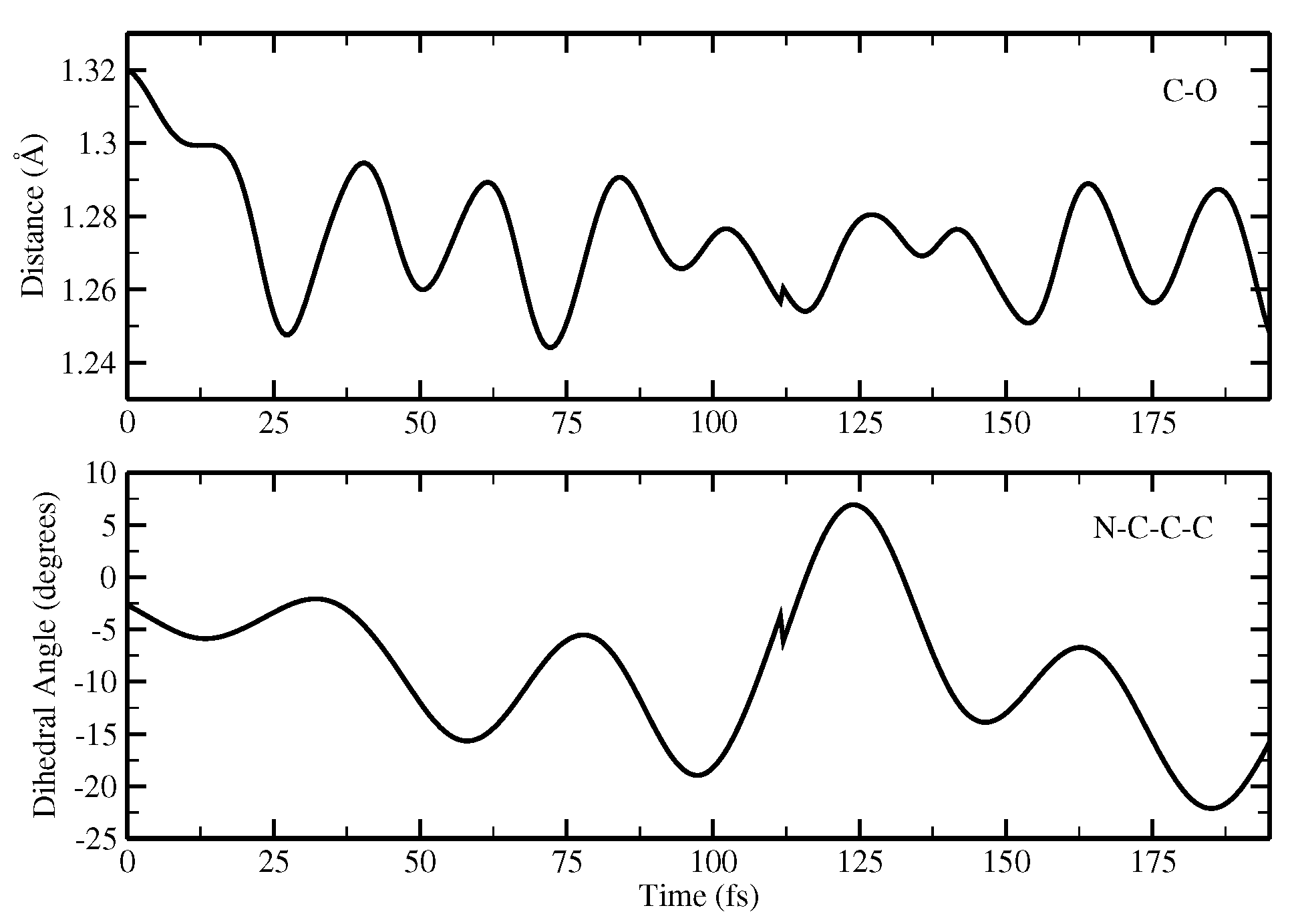
Finally, the ESPT reaction mechanism was investigated by employing ab initio molecular dynamics. Our simulations suggested that the ESPT reaction could take place in a concerted way in a few tens of fs, with two protons moving from the chromophore to Ser148 and from Ser148 to Glu167 at the same time. These results suggest that the proposed hydrogen bond network in the ESPT could be plausible, and that the ultrafast ESPT could take place through indirect proton transfer within a tight hydrogen bond network.






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}