
Gene Therapy in Combination with Nitrogen Scavenger Pretreatment Corrects Biochemical and Behavioral Abnormalities of Infant Citrullinemia Type 1 Mice

 , , , , and
, , , , and 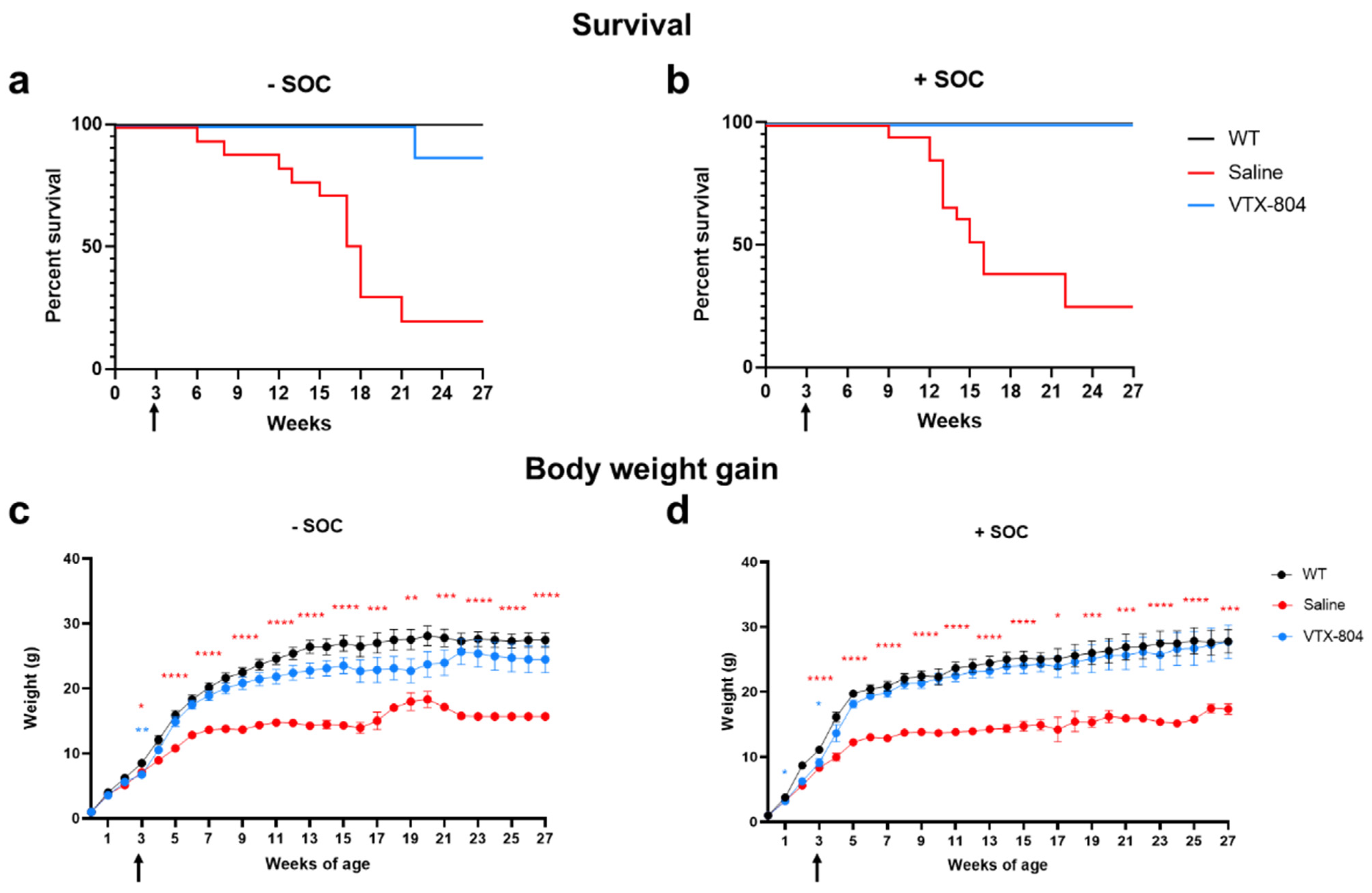{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Combination of VTX-804 Administration with Nitrogen Scavenger Pretreatment Normalizes Survival and Weight Gain of Ass1fold Mice
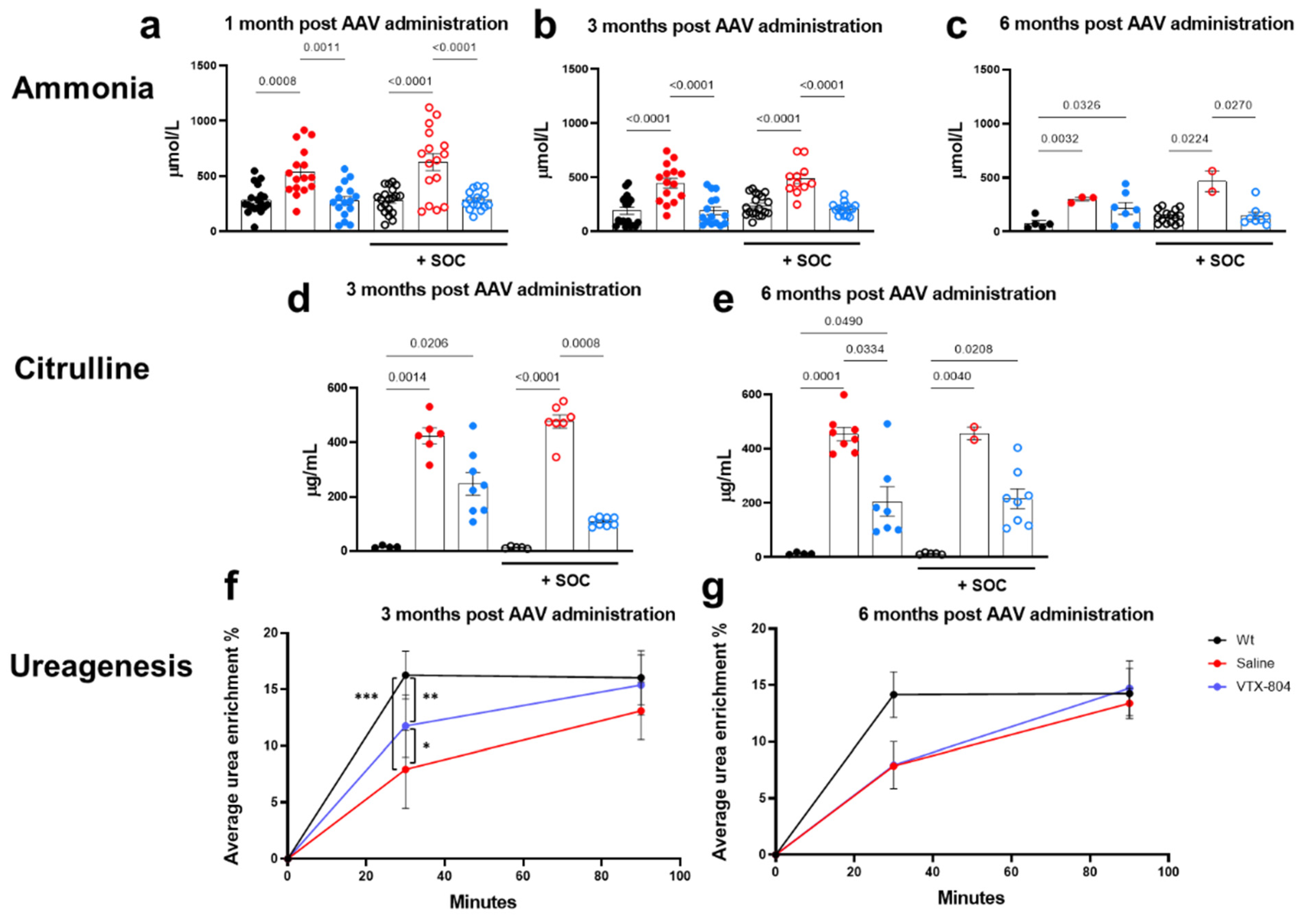
2.2. VTX-804 Activates Ureagenesis in Ass1fold Mice and Improves CTLN1 Serum Biomarkers
2.3. VTX-804 Transduction Levels Diminish with Time
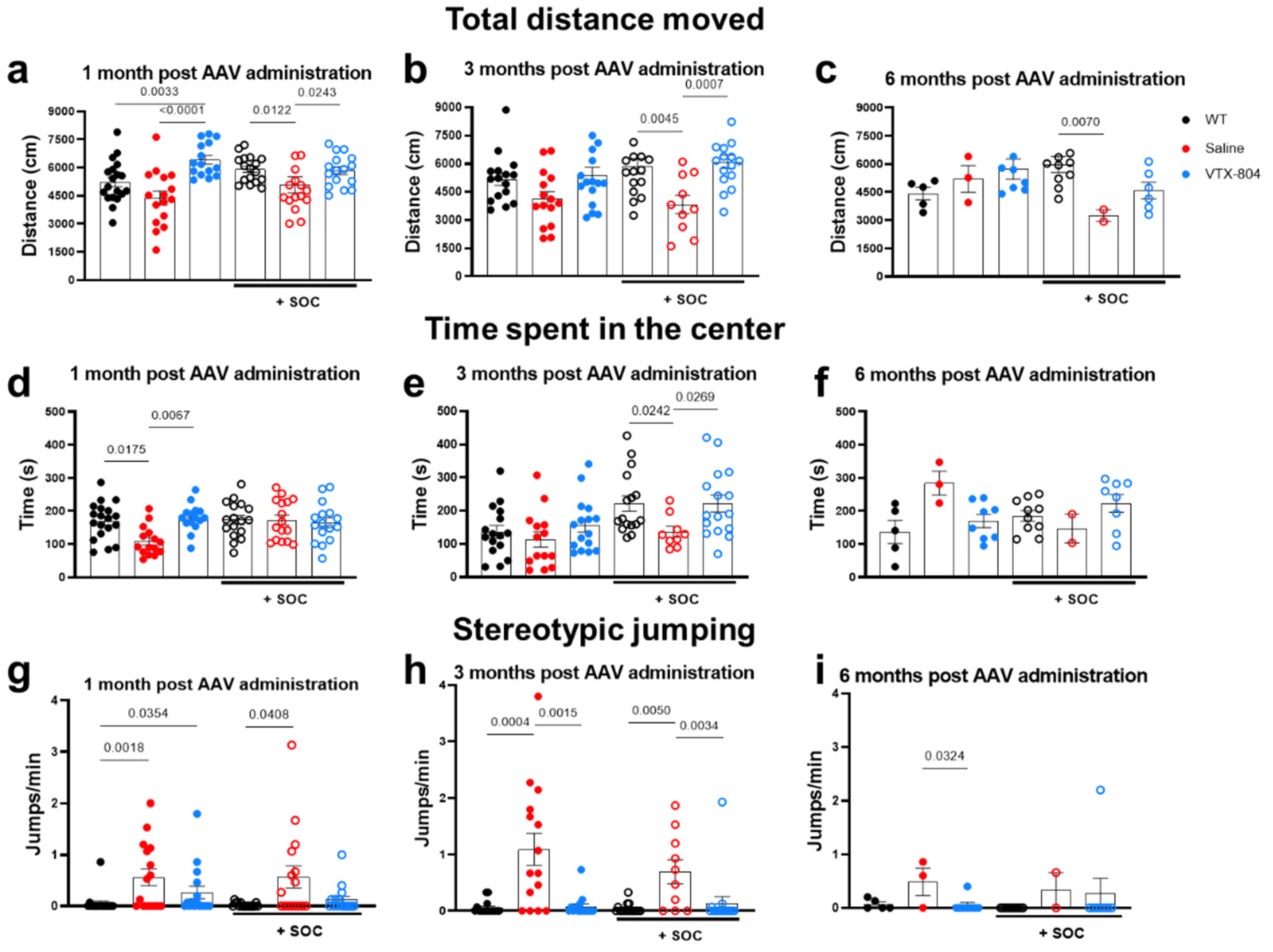
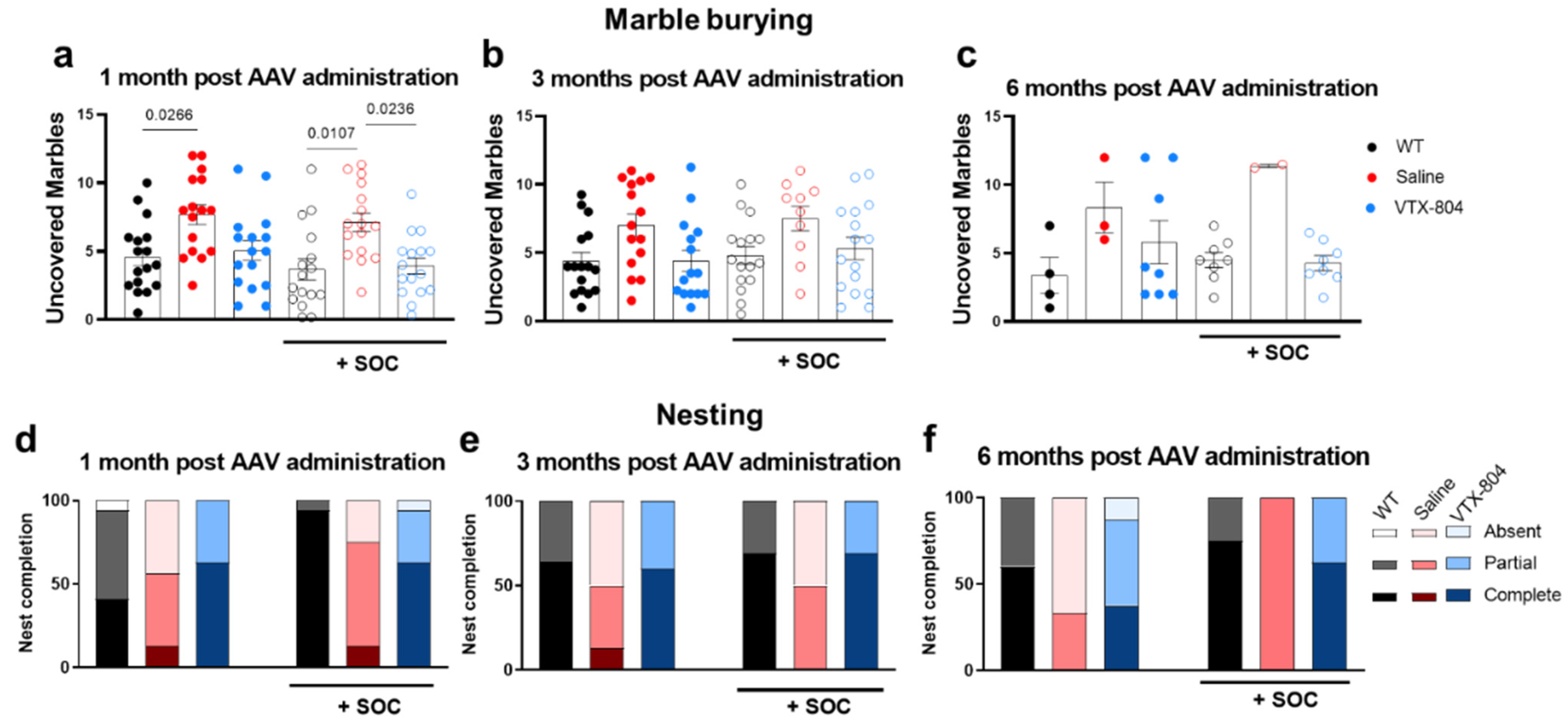
2.4. The Neurological Phenotype Developed by Ass1fold Mice Is Corrected by VTX-804 Treatment
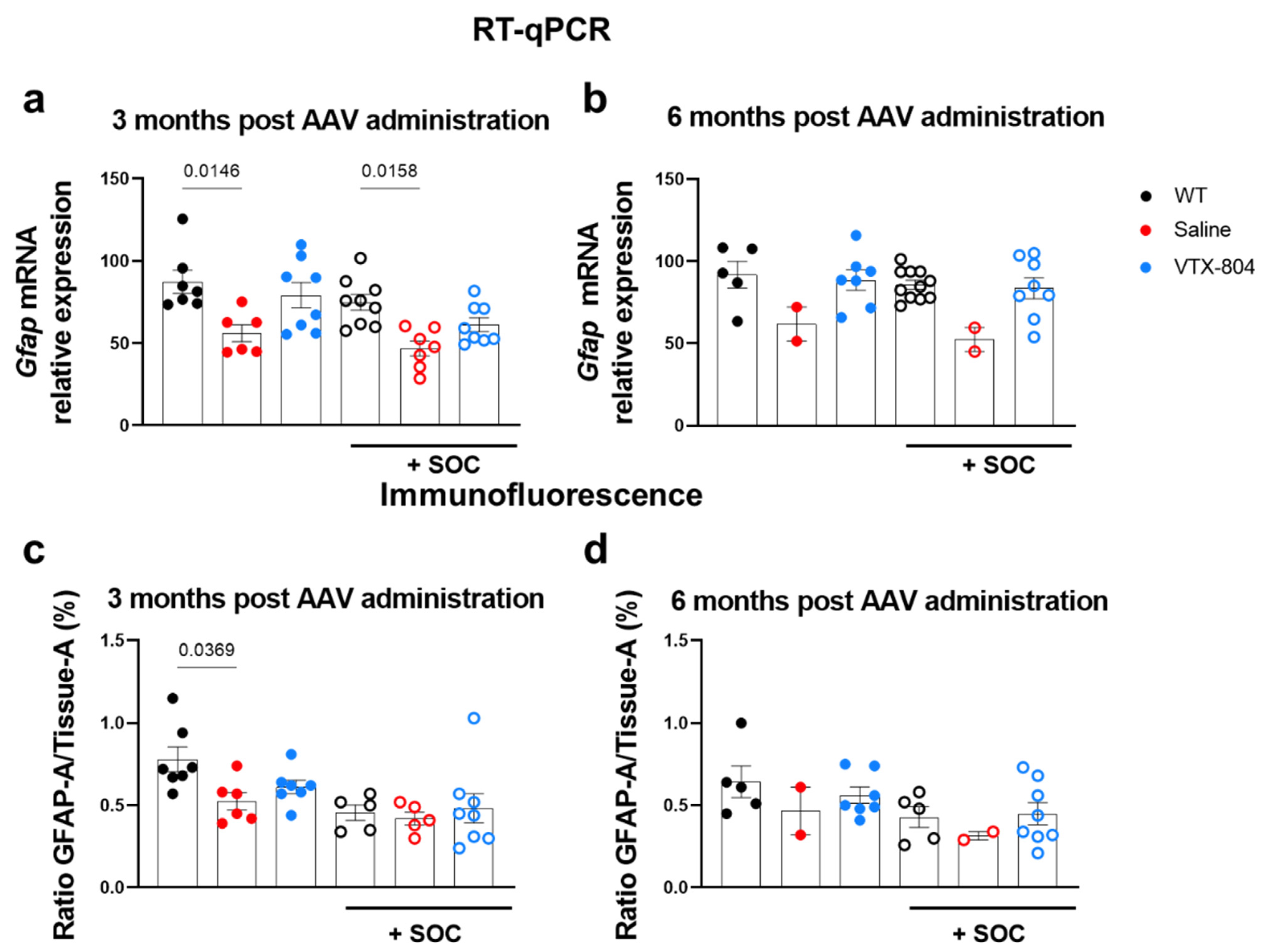
2.5. Histological Brain Alterations Developed by Ass1fold Mice
3. Discussion
4. Materials and Methods
4.1. Cloning and Construction of rAAV Vector
4.2. Production of rAAV Vectors
4.3. Animal Studies
4.4. Analysis of Biochemical Parameters in Serum
4.5. Nucleic Acid Extraction and qPCR
4.6. Ureagenesis
4.7. Behavioural Studies
4.8. Immunofluorescence
4.9. Image Acquisition
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brosnan, M.E.; Brosnan, J.T. Arginine Metabolism: Enzymology, Nutrition, and Clinical Significance. J. Nutr. 2004, 134, 2791S–2795S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, S.; Häberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders—Update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Diez-Fernandez, C.; Rüfenacht, V.; Häberle, J. Mutations in the human Argininosuccinate Synthetase (ASS1) gene, impact on patients, common changes, and structural considerations. Hum. Mutat. 2017, 38, 471–484. [Google Scholar] [CrossRef]
- Burton, B.K. Urea cycle disorders. Liver Dis. Child. Third Ed. 2007, 4, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Walker, V. Ammonia Metabolism and Hyperammonemic Disorders. Adv. Clin. Chem. 2014, 67, 73–150. [Google Scholar] [CrossRef]
- Summar, M.L.; Mew, N.A. Inborn errors of metabolism with hyperammonemia: Urea cycle defects and related disorders. Pediatr. Clin. N. Am. 2018, 65, 231–246. [Google Scholar] [CrossRef]
- Potter, M.A.; Zeesman, S.; Brennan, B.; Kobayashi, K.; Gao, H.-Z.; Tabata, A.; Saheki, T.; Whelan, D.T. Pregnancy in a healthy woman with untreated citrullinemia. Am. J. Med. Genet. 2004, 129, 77–82. [Google Scholar] [CrossRef]
- Posset, R.; Kölker, S.; Gleich, F.; Okun, J.G.; Gropman, A.L.; Nagamani, S.C.; Scharre, S.; Probst, J.; Walter, M.E.; Hoffmann, G.F.; et al. Severity-adjusted evaluation of newborn screening on the metabolic disease course in individuals with cytosolic urea cycle disorders. Mol. Genet. Metab. 2020, 131, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Quinonez, S.C.; Thoene, J.G. Citrullinemia Type I Summary Genetic counseling. GeneReviews 2016, 7, 1–18. [Google Scholar]
- Zielonka, M.; Kölker, S.; Gleich, F.; Stützenberger, N.; Nagamani, S.C.; Gropman, A.L.; Hoffmann, G.F.; Garbade, S.F.; Posset, R.; Urea Cycle Disorders Consortium (UCDC) and the European Registry and Network for Intoxication type Metabolic Diseases (E-IMD) Consortia Study Group; et al. Early prediction of phenotypic severity in Citrullinemia Type 1. Ann. Clin. Transl. Neurol. 2019, 6, 1858–1871. [Google Scholar] [CrossRef] [Green Version]
- Ochoa-Sanchez, R.; Rose, C.F. Pathogenesis of Hepatic Encephalopathy in Chronic Liver Disease. J. Clin. Exp. Hepatol. 2018, 8, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Thrane, V.R.; Thrane, A.S.; Wang, F.; Cotrina, M.L.; Smith, N.A.; Chen, M.; Xu, Q.; Kang, N.; Fujita, T.; Nagelhus, E.A.; et al. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med. 2013, 19, 1643–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.W.; Zieve, L.; Konstantinides, F.N.; Cerra, F.B. Mechanism of arginine protection against ammonia intoxica-tion in the rat. Am. J. Physiol.—Gastrointest. Liver Physiol. 1984, 10, 290–295. [Google Scholar] [CrossRef]
- Posset, R.; Gropman, A.L.; Nagamani, S.C.S.; Burrage, L.C.; Bedoyan, J.K.; Wong, D.; Berry, G.T.; Baumgartner, M.R.; Yudkoff, M.; Zielonka, M.; et al. Impact of Diagnosis and Therapy on Cognitive Function in Urea Cycle Disorders. Ann. Neurol. 2019, 86, 116–128. [Google Scholar] [CrossRef]
- Kido, J.; Matsumoto, S.; Momosaki, K.; Sakamoto, R.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Liver transplantation may pre-vent neurodevelopmental deterioration in high-risk patients with urea cycle disorders. Pediatr. Transplant. 2017, 21, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Truong, B.; Allegri, G.; Liu, X.B.; Burke, K.E.; Zhu, X.; Cederbaum, S.D.; Häberle, J.; Martini, P.G.; Lipshutz, G.S. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 21150–21159. [Google Scholar] [CrossRef]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA therapy for ornithine transcarbamylase deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Spronck, E.A.; Liu, Y.P.; Lubelski, J.; Ehlert, E.; Gielen, S.; Montenegro-Miranda, P.; de Haan, M.; Nijmeijer, B.; Ferreira, V.; Petry, H.; et al. Enhanced factor IX activity following administration of AAV5-R338L “Padua” factor IX versus AAV5 WT human factor IX in NHPs. Mol. Ther.—Methods Clin. Dev. 2019, 15, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2015, 371, 1994–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Zhou, S.; Nakai, H.; Thomas, C.E.; Storm, T.A.; Fuess, S.; Matsushita, T.; Allen, J.; Surosky, R.; Lochrie, M.; et al. Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood 2003, 102, 2412–2419. [Google Scholar] [CrossRef] [PubMed]
- De Sabbata, G.; Boisgerault, F.; Guarnaccia, C.; Iaconcig, A.; Bortolussi, G.; Collaud, F.; Ronzitti, G.; Sola, M.S.; Vidal, P.; Rouillon, J.; et al. Long-term correction of ornithine transcarbamylase deficiency in Spf-Ash mice with a translationally optimized AAV vector. Mol. Ther.—Methods Clin. Dev. 2021, 20, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Morizono, H.; Lin, J.; Bell, P.; Jones, D.; McMenamin, D.; Yu, H.; Batshaw, M.L.; Wilson, J.M. Preclinical evaluation of a clinical candidate AAV8 vector for ornithine transcarbamylase (OTC) deficiency reveals functional enzyme from each persisting vector genome. Mol. Genet. Metab. 2012, 105, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, S.C.; Spinoulas, A.; Carpenter, K.H.; Wilcken, B.; Kuchel, P.W.; Alexander, I.E. AAV2/8-mediated correction of OTC deficiency is robust in adult but not neonatal Spfash mice. Molecular therapy. Mol. Ther. 2009, 17, 1340–1346. [Google Scholar] [CrossRef]
- Lee, E.K.; Hu, C.; Bhargava, R.; Ponnusamy, R.; Park, H.; Novicoff, S.; Rozengurt, N.; Marescau, B.; De Deyn, P.; Stout, D.; et al. AAV-based gene therapy prevents neuropathology and results in normal cognitive development in the hyperargininemic mouse. Gene Ther. 2013, 20, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Nitzahn, M.; Allegri, G.; Khoja, S.; Truong, B.; Makris, G.; Häberle, J.; Lipshutz, G.S. Split AAV-mediated gene therapy restores ureagenesis in a murine model of carbamoyl phosphate synthetase 1 deficiency. Mol. Ther. 2020, 28, 1717–1730. [Google Scholar] [CrossRef]
- Lee, E.K.; Hu, C.; Bhargava, R.; Rozengurt, N.; Stout, D.; Grody, W.W.; Cederbaum, S.D.; Lipshutz, G.S. Long-term survival of the juvenile lethal arginase-deficient mouse with AAV gene therapy. Mol. Ther. 2012, 20, 1844–1851. [Google Scholar] [CrossRef] [Green Version]
- Baruteau, J.; Perocheau, D.P.; Hanley, J.; Lorvellec, M.; Rocha-Ferreira, E.; Karda, R.; Ng, J.; Suff, N.; Diaz, J.A.; Rahim, A.A.; et al. Argininosuccinic aciduria fosters neuronal nitrosative stress reversed by Asl gene transfer. Nat. Commun. 2018, 9, 3505. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Tai, D.S.; Park, H.; Cantero, G.; Chan, E.; Yudkoff, M.; Cederbaum, S.D.; Lipshutz, G.S. Minimal ureagenesis is necessary for survival in the murine model of hyperargininemia treated by AAV-based gene therapy. Gene Ther. 2015, 22, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Ashley, S.N.; Nordin, J.M.L.; Buza, E.L.; Greig, J.A.; Wilson, J.M. Adeno-associated viral gene therapy corrects a mouse model of argininosuccinic aciduria. Mol. Genet. Metab. 2018, 125, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Patejunas, G.; Bradley, A.; Beaudet, A.L.; O’Brien, W.E. Generation of a mouse model for citrullinemia by targeted disruption of the argininosuccinate synthetase gene. Somat. Cell Mol. Genet. 1994, 20, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.J.; Jaubert, J.; Guénet, J.L.; Barnhart, K.F.; Ross-Inta, C.M.; Quintanilla, V.C.; Aubin, I.; Brandon, J.L.; Otto, N.W.; DiGiovanni, J.; et al. Two hypomorphic alleles of mouse Ass1 as a new animal model of citrullinemia type I and other hyperammonemic syndromes. Am. J. Pathol. 2010, 177, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Kok, C.Y.; Cunningham, S.C.; Carpenter, K.H.; Dane, A.P.; Siew, S.M.; Logan, G.J.; Kuchel, P.W.; Alexander, I.E. Adeno-associated virus-mediated rescue of neonatal lethality in argininosuccinate synthetase-deficient mice. Molecular Therapy. Mol. Ther. 2013, 21, 1823–1831. [Google Scholar] [CrossRef] [Green Version]
- Chandler, R.J.; Tarasenko, T.N.; Cusmano-Ozog, K.; Sun, Q.; Sutton, V.R.; Venditti, C.P.; McGuire, P.J. Liver-directed adeno-associated virus serotype 8 gene transfer rescues a lethal murine model of citrullinemia type 1. Gene therapy. Gene Ther. 2013, 20, 1188–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinn, E.; Pacouret, S.; Khaychuk, V.; Turunen, H.T.; Carvalho, L.S.; Andres-Mateos, E.; Shah, S.; Shelke, R.; Maurer, A.C.; Plovie, E.; et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015, 12, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Zabaleta, N.; Hommel, M.; Salas, D.; Gonzalez-Aseguinolaza, G. Genetic-Based Approaches to Inherited Metabolic Liver Diseases. Hum. Gene Ther. 2019, 30, 1190–1203. [Google Scholar] [CrossRef]
- Summar, M.L.; Koelker, S.; Freedenberg, D.; Le Mons, C.; Haberle, J.; Lee, H.S.; Kirmse, B.; Registry, T.E. Members of the Urea Cycle Disorders Consortium. The incidence of urea cycle disorders. Molecular genetics and metabolism. Mol. Genet Metab. 2013, 110, 179–180. [Google Scholar] [CrossRef] [Green Version]
- Häberle, J. Primary hyperammonaemia: Current diagnostic and therapeutic strategies. J. Mother Child 2020, 24, 32–38. [Google Scholar]
- Sonaimuthu, P.; Senkevitch, E.; Haskins, N.; Uapinyoying, P.; McNutt, M.; Morizono, H.; Tuchman, M.; Caldovic, L. Gene delivery corrects N-acetylglutamate synthase deficiency and enables insights in the physiological impact of L-arginine activation of N-acetylglutamate synthase. Sci. Rep. 2021, 11, 3580. [Google Scholar] [CrossRef]
- Peccate, C.; Mollard, A.; Le Hir, M.; Julien, L.; McClorey, G.; Jarmin, S.; Le Heron, A.; Dickson, G.; Benkhelifa-Ziyyat, S.; Piétri-Rouxel, F.; et al. Antisense pre-treatment increases gene therapy efficacy in dystrophic muscles. Hum. Mol. Genet. 2016, 25, 3555–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ros-Gañán, I.; Hommel, M.; Trigueros-Motos, L.; Tamarit, B.; Rodríguez-García, E.; Salas, D.; Pérez, G.; Douar, A.; Combal, J.P.; Benichou, B.; et al. Optimising the IgG-degrading enzyme treatment regimen for enhanced adeno-associated virus transduction in the presence of neutralising antibodies. Clin. Transl. Immunol. 2022, 11, e1375. [Google Scholar] [CrossRef] [PubMed]
- Gropman, A.L.; Prust, M.; Breeden, A.; Fricke, S.; VanMeter, J. Urea cycle defects and hyperammonemia: Effects on functional imaging. Metab. Brain Dis. 2013, 28, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Bélanger, M.; Desjardins, P.; Chatauret, N.; Butterworth, R.F. Loss of expression of glial fibrillary acidic protein in acute hyperammonemia. Neurochem. Int. 2002, 41, 155–160. [Google Scholar] [CrossRef]
- Jaeger, V.; Demorrow, S.; McMillin, M. The direct contribution of astrocytes and microglia to the pathogenesis of hepatic encephalopathy. J. Clin. Transl. Hepatol. 2019, 7, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamiolkowski, D.; on behalf of the E-IMD consortium; Kölker, S.; Glahn, E.M.; Barić, I.; Zeman, J.; Baumgartner, M.R.; Mühlhausen, C.; Garcia-Cazorla, A.; Gleich, F.; et al. Behavioural and emotional problems, intellectual impairment and health-related quality of life in patients with organic acidurias and urea cycle disorders. J. Inherit. Metab. Dis. 2016, 39, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Waisbren, S.E.; Gropman, A.L. Members of the Urea Cycle Disorders Consortium (UCDC), Batshaw ML. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. J. Inherit. Metab. Dis. 2016, 39, 573–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waisbren, S.E.; Members of the Urea Cycle Disorders Consortium; Cuthbertson, D.; Burgard, P.; Holbert, A.; McCarter, R.; Cederbaum, S. Biochemical markers and neuropsychological functioning in distal urea cycle disorders. J. Inherit. Metab. Dis. 2018, 41, 657–667. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Stefanatos, A.K.; Kok, T.M.Y.; Ozturk-Hismi, B. Neuropsychological attributes of urea cycle disorders: A systematic review of the literature. J. Inherit. Metab. Dis. 2019, 42, 1176–1191. [Google Scholar] [CrossRef] [Green Version]
- Baruteau, J.; Diez-Fernandez, C.; Lerner, S.; Ranucci, G.; Gissen, P.; Dionisi-Vici, C.; Nagamani, S.; Erez, A.; Häberle, J. Argininosuccinic aciduria: Recent pathophysiological insights and therapeutic prospects. Journal of Inherited Metabolic Disease. J. Inherit. Metab. Dis. 2019, 42, 1147–1161. [Google Scholar] [CrossRef] [Green Version]
- Kramer, M.G.; Barajas, M.; Razquin, N.; Berraondo, P.; Rodrigo, M.; Wu, C.; Qian, C.; Fortes, P.; Prieto, J. In vitro and in vivo comparative study of chimeric liver-specific promoters. Mol. Ther. 2003, 7, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Grumbach, E.S.; Wheat, T.E.; Mazzeo, J.R. A Novel Method for the Analysis of Amino Acids. Available online: www.waters.com (accessed on 20 November 2022).
- Allegri, G.; Deplazes, S.; Grisch-Chan, H.M.; Mathis, D.; Fingerhut, R.; Häberle, J.; Thöny, B. A simple dried blood spot-method for in vivo measurement of ureagenesis by gas chromatography–mass spectrometry using stable isotopes. Clin. Chim. Acta 2017, 464, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Mora-Jimenez, L.; Valencia, M.; Sanchez-Carpintero, R.; Tønnesen, J.; Fadila, S.; Rubinstein, M.; Gonzalez-Aparicio, M.; Bunuales, M.; Fernandez-Pierola, E.; Nicolas, M.J.; et al. Transfer of SCN1A to the brain of adolescent mouse model of Dravet syndrome improves epileptic, motor, and behavioral manifestations. Mol. Ther.—Nucleic Acids 2021, 25, 585–602. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazo, A.; Lantero, A.; Mauleón, I.; Neri, L.; Poms, M.; Häberle, J.; Ricobaraza, A.; Bénichou, B.; Combal, J.-P.; Gonzalez-Aseguinolaza, G.; et al. Gene Therapy in Combination with Nitrogen Scavenger Pretreatment Corrects Biochemical and Behavioral Abnormalities of Infant Citrullinemia Type 1 Mice. Int. J. Mol. Sci. 2022, 23, 14940. https://doi.org/10.3390/ijms232314940
Bazo A, Lantero A, Mauleón I, Neri L, Poms M, Häberle J, Ricobaraza A, Bénichou B, Combal J-P, Gonzalez-Aseguinolaza G, et al. Gene Therapy in Combination with Nitrogen Scavenger Pretreatment Corrects Biochemical and Behavioral Abnormalities of Infant Citrullinemia Type 1 Mice. International Journal of Molecular Sciences. 2022; 23(23):14940. https://doi.org/10.3390/ijms232314940
Chicago/Turabian StyleBazo, Andrea, Aquilino Lantero, Itsaso Mauleón, Leire Neri, Martin Poms, Johannes Häberle, Ana Ricobaraza, Bernard Bénichou, Jean-Philippe Combal, Gloria Gonzalez-Aseguinolaza, and et al. 2022. "Gene Therapy in Combination with Nitrogen Scavenger Pretreatment Corrects Biochemical and Behavioral Abnormalities of Infant Citrullinemia Type 1 Mice" International Journal of Molecular Sciences 23, no. 23: 14940. https://doi.org/10.3390/ijms232314940