

Conformational Structures of Neutral and Cationic Pivaldehyde Revealed by IR-Resonant VUV-MATI Mass Spectroscopy

Abstract
:1. Introduction
2. Results and Discussion
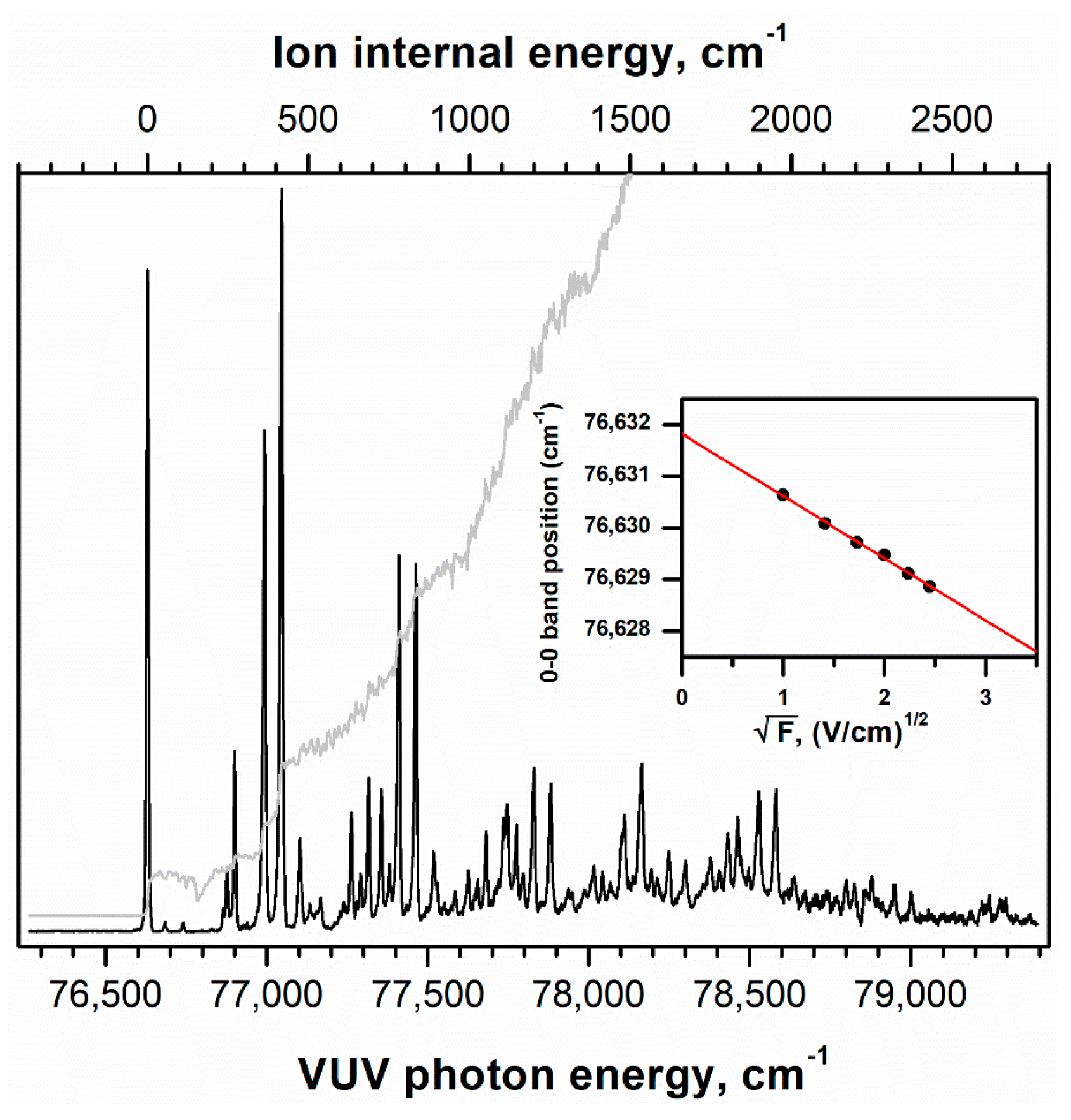
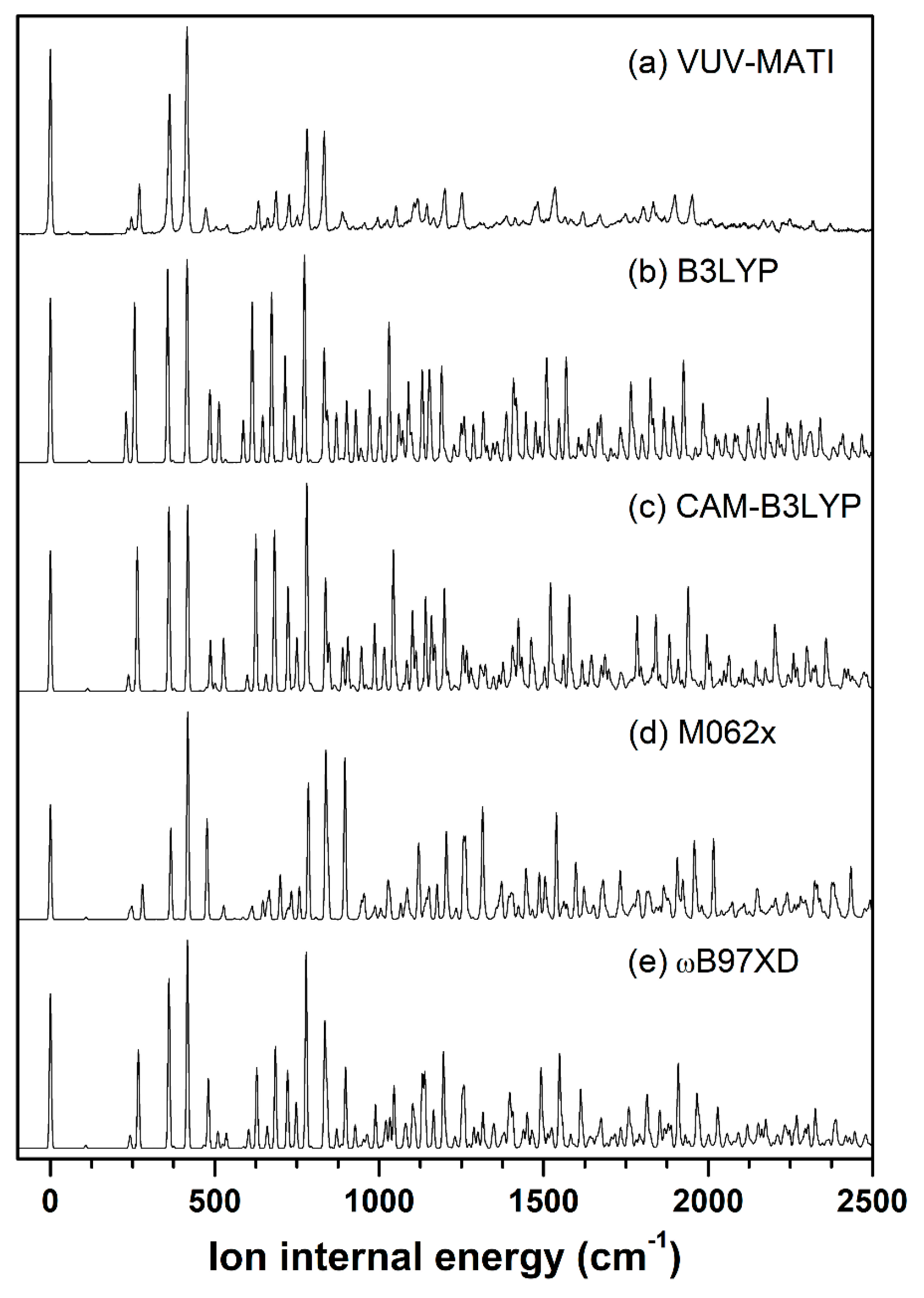
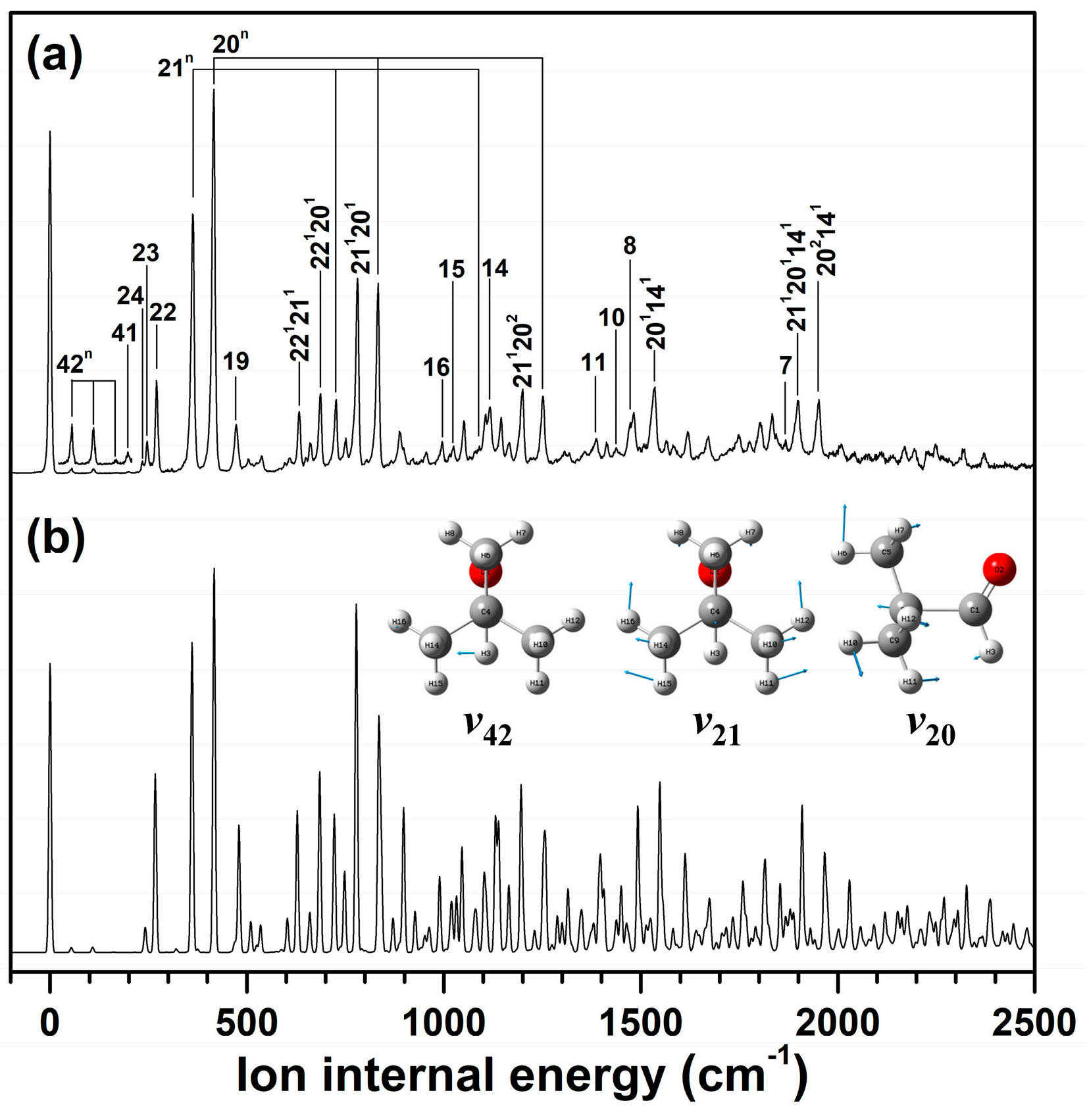
2.1. Conformer-Specific Photoionization of Pivaldehyde
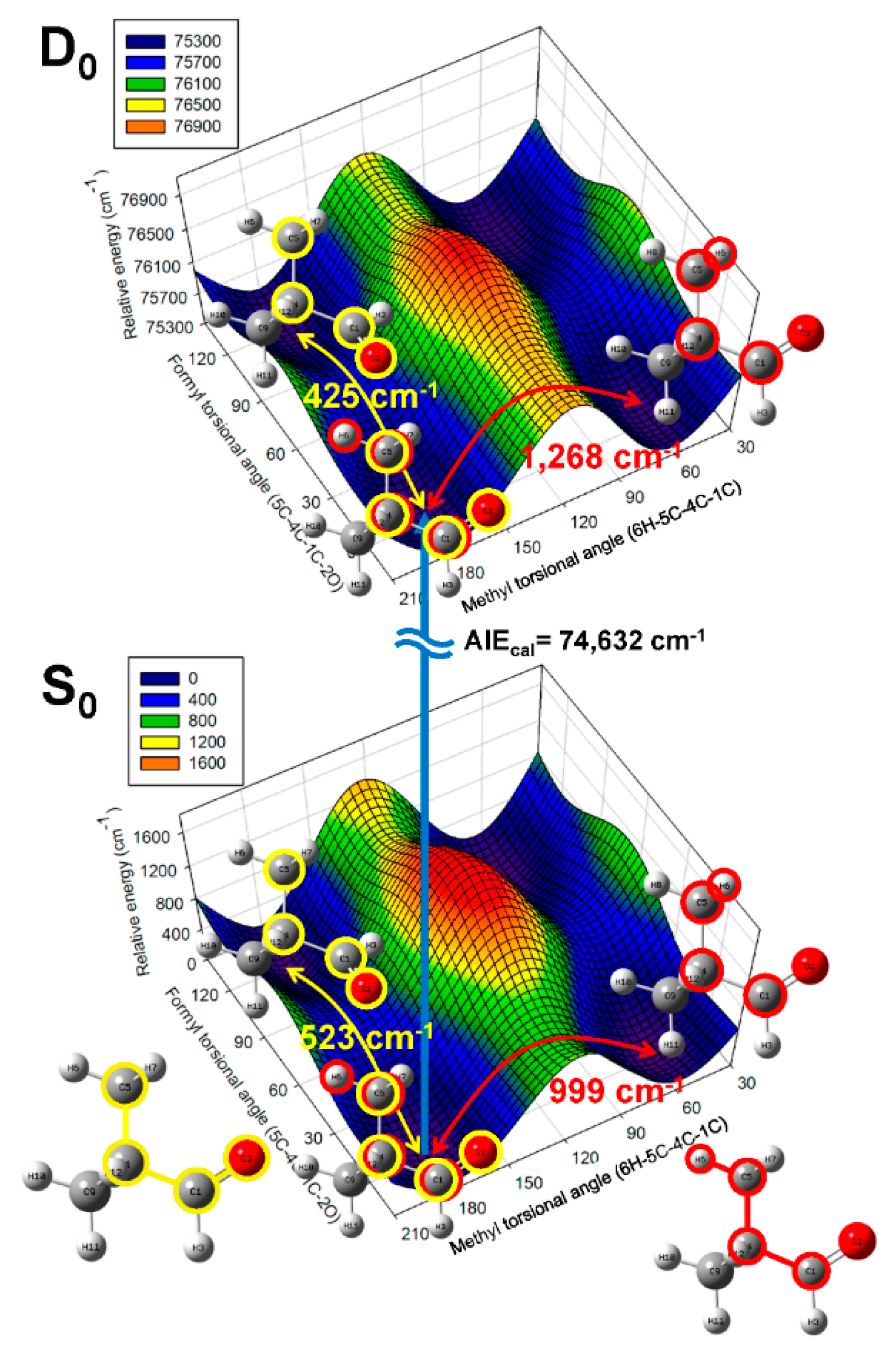
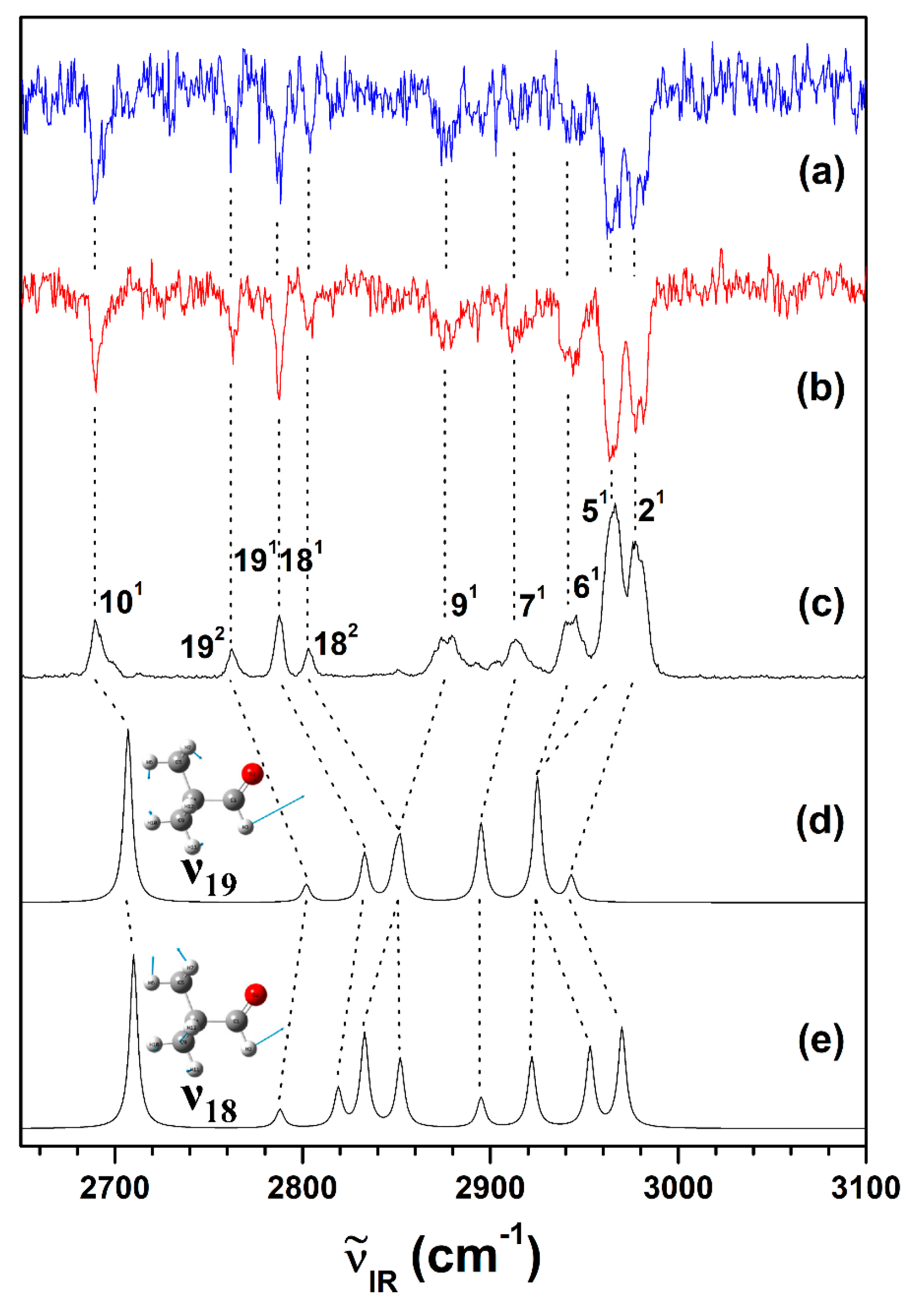
2.2. Conformational Structure of Neutral Pivaldehyde
2.3. Cationic Structure of Pivaldehyde
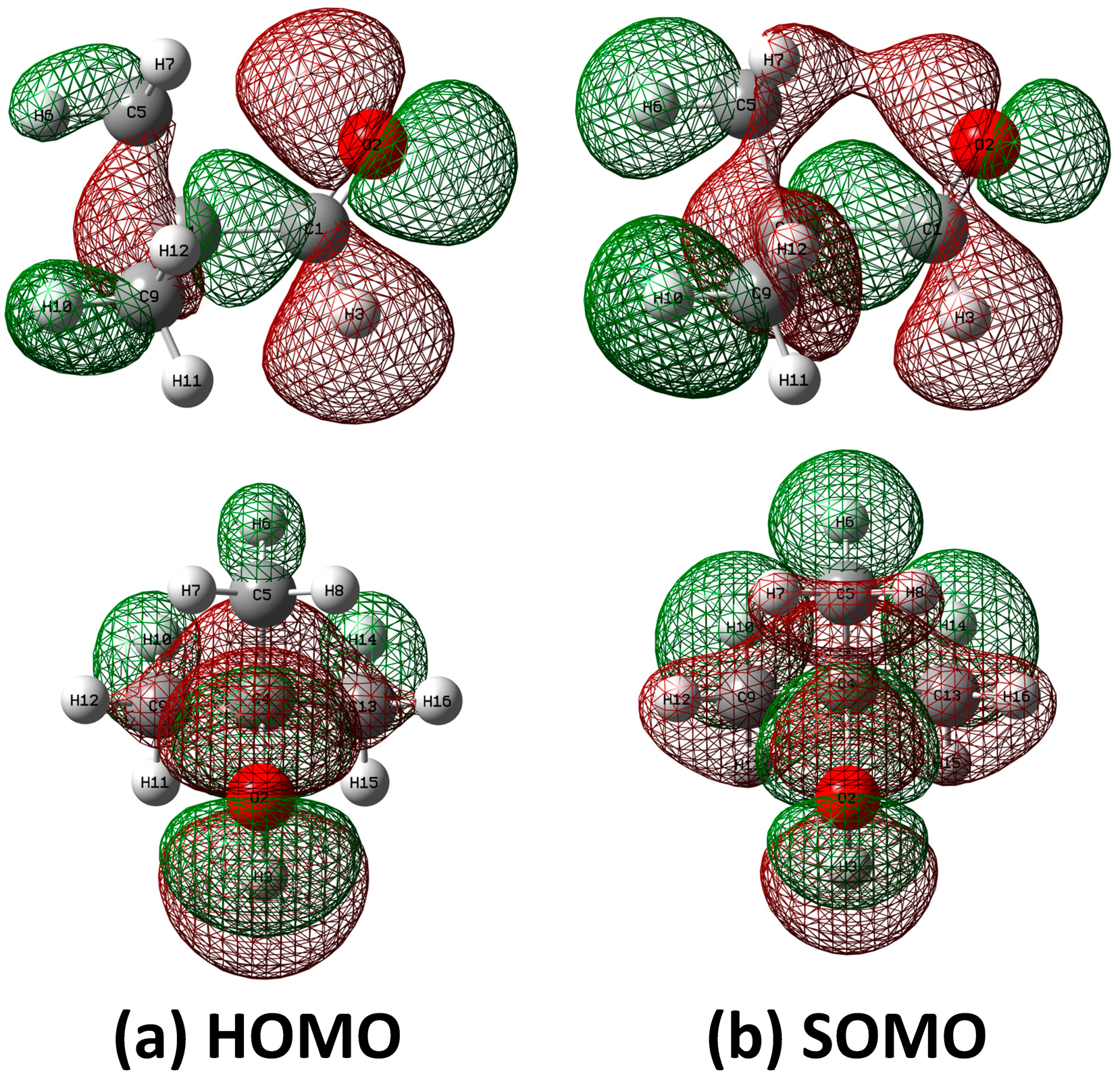
2.4. HOMO and SOMO of Pivaldehyde
3. Materials and Methods
3.1. Experimental Methods
3.2. Theoretical Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crim, F.F. Bond-Selected Chemistry: Vibrational state control of photodissociation and bimolecular reaction. J. Phys. Chem. 1996, 100, 12725–12734. [Google Scholar] [CrossRef]
- Khriachtchev, L.; Maҫôas, E.; Pettersson, M.; Räsänen, M. Conformational memory in photodissociation of formic acid. J. Am. Chem. Soc. 2002, 124, 10994–10995. [Google Scholar] [CrossRef] [PubMed]
- Dian, B.C.; Longarte, A.; Zwier, T.S. Conformational dynamics in a dipeptide after single-mode vibrational excitation. Science 2002, 296, 2369–2373. [Google Scholar] [CrossRef]
- Park, S.T.; Kim, S.K.; Kim, M.S. Observation of conformation-specific pathways in the photodissociation of 1-iodopropane ions. Nature 2002, 415, 306–308. [Google Scholar] [CrossRef]
- Kim, M.H.; Shen, L.; Tao, H.L.; Martinez, T.J.; Suits, A.G. Conformationally controlled chemistry: Excited-state dynamics dictate ground-state reaction. Science 2007, 315, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.R. Free Jet Sources. In Atomic and Molecular Beam Methods; Scoles, G., Ed.; Oxford University Press: New York, NY, USA, 1988; Volume 1, pp. 14–53. [Google Scholar]
- Park, S.M.; Lee, Y.R.; Kang, D.W.; Kim, H.L.; Kwon, C.H. Conformational structures of the tetrahydrofuran cation determined using one-photon mass-analyzed threshold ionization spectroscopy. Phys. Chem. Chem. Phys. 2017, 19, 30362–30369. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kim, M.H.; Kim, H.L.; Kwon, C.H. Conformer-specific photoionization and conformational stabilities of isobutanal revealed by one-photon mass-analyzed threshold ionization (MATI) spectroscopy. J. Chem. Phys. 2018, 149, 174302. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, H.L.; Kwon, C.H. Identification and composition of conformational isomers and their cations in crotonaldehyde by VUV-MATI spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 27162–27168. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kim, K.E.; Kim, M.H.; Kim, H.L.; Kwon, C.H. Formyl torsion and cationic structure of gauche conformer in isobutanal by conformer-specific VUV-MATI spectroscopy and Franck-Condon fitting. J. Chem. Phys. 2019, 151, 014304. [Google Scholar] [CrossRef]
- Eom, S.Y.; Lee, Y.R.; Kwon, C.H. Accurate conformational stability and cationic structure of piperidine determined by conformer-specific VUV-MATI spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 6184–6191. [Google Scholar] [CrossRef]
- Park, S.M.; Choi, J.; Kim, H.L.; Kwon, C.H. Conformer-specific VUV-MATI spectroscopy of methyl vinyl ketone: Stabilities and cationic structures of the s-trans and s-cis conformers. Phys. Chem. Chem. Phys. 2020, 22, 28383–28392. [Google Scholar] [CrossRef]
- Eom, S.Y.; Kang, D.W.; Kwon, C.H. Conformational structure of cationic tetrahydropyran by one-photon vacuum ultraviolet mass-analyzed threshold ionization spectroscopy. Phys. Chem. Chem. Phys. 2021, 23, 1414–1423. [Google Scholar] [CrossRef]
- Lipert, R.J.; Colson, S.D. Persistent spectral hole burning of molecular clusters in a supersonic jet. J. Phys. Chem. 1989, 93, 3894–3896. [Google Scholar] [CrossRef]
- Riehn, C.; Lahmann, C.; Wassermann, B.; Brutschy, B. IR depletion spectroscopy. A method for characterizing a microsolvation environment. Chem. Phys. Lett. 1992, 197, 443–450. [Google Scholar] [CrossRef]
- Ebata, T.; Fujii, A.; Mikami, N. Vibrational spectroscopy of small-sized hydrogen-bonded clusters and their ions. Int. Rev. Phys. Chem. 1998, 17, 331–361. [Google Scholar] [CrossRef]
- Bakker, J.M.; Mac Aleese, L.; Meijer, G.; Helden, G. Fingerprint IR spectroscopy to probe amino acid conformations in the gas phase. Phys. Rev. Lett. 2003, 91, 203003. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.M.; Choi, D.H.; Heo, J.; Kim, N.J.; Kim, S.K. Ultraviolet–Ultraviolet hole burning spectroscopy in a quadrupole ion trap: Dibenzo [18] crown-6 complexes with alkali metal cations. Angew. Chem. Int. Ed. 2012, 51, 7297–7300. [Google Scholar] [CrossRef]
- Min, A.; Ahn, A.; Moon, C.J.; Lee, J.H.; Seong, Y.G.; Kim, S.K.; Choi, M.Y. Conformational structures of jet-cooled acetaminophen-water clusters: A gas phase spectroscopic and computational study. Phys. Chem. Chem. Phys. 2017, 19, 4840–4848. [Google Scholar] [CrossRef]
- Woo, H.K.; Wang, P.; Lau, K.-C.; Xing, X.; Chang, C.; Ng, C.Y. State-selected and state-to-state photoionization study of trichloroethene using the two-color infrared-vacuum ultraviolet scheme. J. Chem. Phys. 2003, 119, 9333–9336. [Google Scholar] [CrossRef]
- Matsuda, Y.; Mikami, N.; Fujii, A. Vibrational spectroscopy of size-selected neutral and cationic clusters combined with vacuum-ultraviolet one-photon ionization detection. Phys. Chem. Chem. Phys. 2009, 11, 1279–1290. [Google Scholar] [CrossRef]
- Han, H.; Camacho, C.; Witek, H.A.; Lee, Y. Infrared absorption of methanol clusters (CH3OH)n with n = 2–6 recorded with a time-of flight mass spectrometer using infrared depletion and vacuum-ultraviolet ionization. J. Chem. Phys. 2011, 134, 144309. [Google Scholar] [CrossRef]
- Park, S.M.; Kwon, C.H. Development and verification of conformer-specific vibrational spectroscopy. J. Phys. Chem. A 2021, 125, 9251–9258. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Kwon, C.H. Identification of individual conformers in C4H6O isomers using conformer-specific vibrational spectroscopy. RSC Adv. 2021, 11, 38240–38246. [Google Scholar] [CrossRef] [PubMed]
- Vasyunin, A.I.; Herbst, E. Reactive desorption and radiative association as possible drivers of complex molecule formation in the cold interstellar medium. Astrophys. J. 2013, 769, 34. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, R.I.; Maity, S.; Jones, B.M. Infrared and reflectron time-of-flight mass spectroscopic analysis of methane (CH4)–carbon monoxide (CO) ices exposed to ionization radiation—Toward the formation of carbonyl bearing molecules in extraterrestrial ices. Phys. Chem. Chem. Phys. 2014, 16, 3399–3424. [Google Scholar] [CrossRef] [PubMed]
- McGuire, B. 2018 Census of interstellar, circumstellar, extragalactic, protoplanetary disk, and exoplanetary molecules. Astrophys. J. Suppl. Ser. 2018, 239, 48. [Google Scholar] [CrossRef]
- Kim, M.H.; Shen, L.; Suits, A.G. Photoelectron imaging of propanal by resonant multiphoton ionization via the 3s Rydberg state. Phys. Chem. Chem. Phys. 2006, 8, 2933–2939. [Google Scholar] [CrossRef]
- Choi, S.; Kang, T.Y.; Choi, K.; Han, S.; Ahn, D.; Baek, S.J.; Kim, S.K. Ionization spectroscopy of conformational isomers of propanal: The origin of the conformational preference. J. Phys. Chem. A 2008, 112, 5060–5063. [Google Scholar] [CrossRef]
- Shen, L.; Singh, P.C.; Kim, M.; Zhang, B.; Suits, A.G. Ion and electron imaging study of isobutanal photoionization dynamics. J. Phys. Chem. A 2009, 113, 68–74. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kwon, C.H. Innovative mass spectrometer for high resolution ion spectroscopy. J. Chem. Phys. 2021, 155, 164203. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E. Vacuum Ultraviolet Mass-Analyzed Threshold Ionization (VUV-MATI) Spectroscopy of Trimethylacetaldehyde. Master’s Thesis, Ewha University, Seoul, Republic of Korea, 2017. [Google Scholar]
- Ronn, A.M.; Woods, R.C., III. Microwave spectrum of pivaldehyde. J. Chem. Phys. 1966, 45, 3831–3836. [Google Scholar] [CrossRef]
- Crowder, G.A. Vibrational spectra of 2,2-dimethylpropanal. J. Chem. Soc. Perkin Trans. II 1973, 2, 1241–1244. [Google Scholar] [CrossRef]
- Durig, J.R.; Kenton, R.; Phan, H.V.; Little, T.S. Far-infrared spectra and barriers to internal rotation of pivaldehyde, pivaloyl fluoride, pivaloyl chloride and pinacolone. J. Mol. Struct. 1991, 247, 237–251. [Google Scholar] [CrossRef]
- Cox, A.P.; Couch, A.D.; Hillig II, K.W.; LaBarge, M.S.; Kuczkowski, R.L. Microwave spectrum, molecular structure and dipole moment of pivalaldehyde. J. Chem. Soc. Faraday Trans. 1991, 87, 2689–2698. [Google Scholar] [CrossRef]
- Kim, M.C.; Sim, E.; Burke, K. Understanding and reducing errors in density functional calculations. Phys. Rev. Lett. 2013, 111, 073003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrein, A.; Hollenstein, H.; Quack, M.; Zenobi, R.; Segall, J.; Zare, R.N. Fermi resonance structure in the CH vibrational overtones of CD3CHO. J. Chem. Phys. 1989, 90, 3944–3951. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.R.; Kwon, C.H. Determination of highest occupied molecular orbital and cationic structures of cyclopentanone using high-resolution vacuum ultraviolet mass-analyzed threshold ionization mass spectrophotometry. Bull. Korean Chem. Soc. 2022, 43, 636–644. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sinha, P.; Boesch, S.E.; Gu, C.; Wheeler, R.A.; Wilson, A.K. Harmonic vibrational frequencies: Scaling factors for HF, B3LYP, and MP2 methods in combination with correlation consistent basis sets. J. Phys. Chem. A 2004, 108, 9213–9217. [Google Scholar] [CrossRef]
- Merrick, J.P.; Moran, D.; Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode a | S0 (C1) | D0 (CS) | ||||||
|---|---|---|---|---|---|---|---|---|
| VUV-MATI | FC (CS→) d | FC (C1→) c | ||||||
| Exp b | Cal c | Freq | Int e | Freq | Int e | Freq | Int e | |
| Fundamentals | ||||||||
| 421 | 70 | 77 | 55 | 0.013 | 54 | 54 | 0.014 | |
| 411 | 222 | 196 | 199 | 0.004 | 195 | 195 | 1 × 10−4 | |
| 391 | 330 | 322 | 309 | 0.012 | 308 | 308 | 5 × 10−4 | |
| 241 | 267 | 250 | 235 | 0.031 | 239 | 0.012 | 239 | 0.013 |
| 231 | 278 | 275 | 246 | 0.082 | 242 | 0.059 | 242 | 0.058 |
| 221 | 348 | 346 | 270 | 0.242 | 267 | 0.475 | 267 | 0.472 |
| 211 | 389 | 385 | 363 | 0.676 | 361 | 0.814 | 361 | 0.815 |
| 201 | 593 | 590 | 416 | 1.000 | 417 | 1.000 | 417 | 1.000 |
| 191 | 764 | 759 | 473 | 0.126 | 480 | 0.330 | 480 | 0.331 |
| 161 | 1041 | 1038 | 996 | 0.081 | 989 | 0.069 | 989 | 0.069 |
| 151 | 1217 | 1222 | 1025 | 0.070 | 1032 | 0.146 | 1032 | 0.147 |
| 141 | 1277 | 1279 | 1117 | 0.171 | 1131 | 0.347 | 1131 | 0.347 |
| 111 | 1406 | 1410 | 1387 | 0.089 | 1398 | 0.006 | 1398 | 0.006 |
| 101 | 1457 | 1453 | 1437 | 0.067 | 1435 | 0.003 | 1435 | 0.003 |
| 81 | 1484 | 1489 | 1473 | 0.132 | 1483 | 0.002 | 1483 | 0.002 |
| 71 | 1774 | 1805 | 1867 | 0.086 | 1888 | 0.075 | 1888 | 0.074 |
| Overtones and combinations | ||||||||
| 422 | 130 | 154 | 110 | 0.009 | 108 | 0.016 | 108 | 0.013 |
| 423 | 166 | 0.001 | 162 | 162 | 0.001 | |||
| 241221 | 504 | 0.036 | 506 | 0.001 | 506 | 0.001 | ||
| 222 | 538 | 0.045 | 535 | 0.069 | 535 | 0.067 | ||
| 241211 | 596 | 0.027 | 600 | 0.007 | 600 | 0.008 | ||
| 231211 | 610 | 0.039 | 603 | 0.086 | 603 | 0.085 | ||
| 221211 | 632 | 0.156 | 628 | 0.375 | 628 | 0.373 | ||
| 241201 | 650 | 0.039 | 656 | 0.011 | 656 | 0.013 | ||
| 231201 | 662 | 0.076 | 660 | 0.100 | 660 | 0.099 | ||
| 221201 | 685 | 0.196 | 685 | 0.476 | 685 | 0.472 | ||
| 212 | 726 | 0.185 | 722 | 0.339 | 722 | 0.340 | ||
| 221191 | 750 | 0.081 | 748 | 0.213 | 748 | 0.211 | ||
| 211201 | 780 | 0.507 | 778 | 0.882 | 778 | 0.882 | ||
| 202 | 833 | 0.494 | 835 | 0.579 | 835 | 0.578 | ||
| 201191 | 888 | 0.105 | 898 | 0.351 | 898 | 0.352 | ||
| 222201 | 955 | 0.054 | 952 | 0.036 | 952 | 0.035 | ||
| 222191 | 1015 | 0.050 | 1015 | 0.033 | 1015 | 0.032 | ||
| 221211201 | 1051 | 0.135 | 1046 | 0.274 | 1046 | 0.272 | ||
| 213 | 1090 | 0.070 | 1082 | 0.065 | 1082 | 0.066 | ||
| 221211191 | 1106 | 0.153 | 1108 | 0.115 | 1108 | 0.115 | ||
| 212201 | 1145 | 0.144 | 1139 | 0.280 | 1139 | 0.280 | ||
| 211202 | 1200 | 0.220 | 1196 | 0.401 | 1196 | 0.400 | ||
| 203 | 1251 | 0.199 | 1252 | 0.191 | 1252 | 0.191 | ||
| 202191 | 1306 | 0.057 | 1315 | 0.155 | 1315 | 0.155 | ||
| 211161 | 1357 | 0.057 | 1350 | 0.060 | 1450 | 0.060 | ||
| 201161 | 1413 | 0.008 | 1406 | 0.084 | 1406 | 0.084 | ||
| 211141 | 1482 | 0.157 | 1492 | 0.349 | 1492 | 0.349 | ||
| 191151 | 1496 | 0.068 | 1513 | 0.062 | 1513 | 0.063 | ||
| 201141 | 1536 | 0.225 | 1548 | 0.435 | 1548 | 0.435 | ||
| 212201191 | 1619 | 0.108 | 1619 | 0.028 | 1619 | 0.028 | ||
| 211202191 | 1671 | 0.095 | 1676 | 0.056 | 1676 | 0.056 | ||
| 221211141 | 1748 | 0.099 | 1759 | 0.151 | 1759 | 0.150 | ||
| 231201141 | 1776 | 0.082 | 1791 | 0.051 | 1791 | 0.051 | ||
| 221201141 | 1803 | 0.131 | 1816 | 0.196 | 1816 | 0.194 | ||
| 202161 | 1833 | 0.154 | 1824 | 0.045 | 1824 | 0.044 | ||
| 212141 | 1843 | 0.102 | 1853 | 0.134 | 1853 | 0.134 | ||
| 211201141 | 1899 | 0.188 | 1909 | 0.356 | 1909 | 0.356 | ||
| 202141 | 1952 | 0.191 | 1966 | 0.239 | 1966 | 0.238 | ||
| 201191141 | 2008 | 0.075 | 2029 | 0.150 | 2029 | 0.150 | ||
| 231202141 | 2195 | 0.064 | 2208 | 0.036 | 2208 | 0.036 | ||
| 203161 | 2248 | 0.075 | 2241 | 0.013 | 2241 | 0.013 | ||
| 211202141 | 2320 | 0.061 | 2327 | 0.151 | 2327 | 0.150 | ||
| 14181 | 2591 | 0.041 | 2614 | 0.001 | 2614 | 0.001 | ||
| 191151141 | 2614 | 0.049 | 2644 | 0.035 | 2644 | 0.035 | ||
| S0 | D0 | |||
|---|---|---|---|---|
| Exp a | Cal (Cs) b | Cal (C1) c | Cal (Cs) b | |
| Bond length (Å) | ||||
| 1C–2O | 1.206 | 1.199 | 1.199 | 1.159 (−0.040) d |
| 1C–4C | 1.516 | 1.516 | 1.516 | 1.724 (0.208) |
| 4C–5C | 1.537 | 1.523 | 1.523 | 1.505 (−0.018) |
| 4C–9C (4C–13C) | 1.537 | 1.535 | 1.535 | 1.515 (−0.020) |
| 1C–3H | 1.130 | 1.113 | 1.113 | 1.103 (−0.010) |
| 5C–6H | 1.118 | 1.091 | 1.091 | 1.093 (0.002) |
| 5C–7H (5C–8H) | 1.118 | 1.089 | 1.089 | 1.088 (−0.001) |
| 9C–10H (13C–14H) | 1.118 | 1.091 | 1.091 | 1.095 (0.004) |
| 9C–11H (13C–15H) | 1.118 | 1.092 | 1.092 | 1.089 (−0.003) |
| 9C–12H (13C–16H) | 1.118 | 1.091 | 1.091 | 1.089 (−0.002) |
| Bond angle (°) | ||||
| 2O–1C–4C | 126.0 | 125.9 | 125.9 | 121.0 (−4.9) |
| 1C–4C–5C | 110.5 | 110.9 | 110.8 | 108.3 (−2.5) |
| 1C–4C–9C (1C–4C–13C) | 107.4 | 107.1 | 106.9 | 100.5 (−6.4) |
| 4C–1C–3H | 113.0 | 113.8 | 113.8 | 110.6 (−3.2) |
| Dihedral angle (°) | ||||
| 2O–1C–4C–5C | 2.1 | 0.0 | 2.1 | 0.0 (−2.1) |
| Mode (C1) a | IR(CS) b | Cal(CS) c (Har) | Cal(CS) d (Anhar) | Cal(C1) e (Anhar) | IR-Resonant VUV-PI | IR-Dip VUV-MATI | Mode Description f |
|---|---|---|---|---|---|---|---|
| 101 | 2696 | 2727 | 2707 | 2710 | 2689 | 2689 | Cformyl-H stretch |
| 192 | 2768 | 2695 | 2802 | 2788 | 2762 | 2762 | Overtone of Cformyl-H bending |
| 191181 | 2791 | 2722 | 2833 | 2819 | 2788 | 2788 | Combination of bending |
| 182 | 2803 | 2750 | 2849 | 2833 | 2803 | 2803 | Overtone of Cformyl-H bending |
| 91 | 2876 | 2894 | 2852 | 2852 | 2876 | 2876 | Out-of-plane (CH3)2 asym. stretch |
| 71 | 2913 | 2904 | 2895 | 2895 | 2913 | 2913 | In-plane CH3 sym. stretch |
| 61 | 2922 | 2943 | 2943 | Out-of-plane (CH3)2 asym. stretch | |||
| 51 | 2969 | 2964 | 2925 | 2953 | 2964 | 2964 | Out-of-plane (CH3)2 sym. stretch |
| 21 | 2980 | 2976 | 2943 | 2970 | 2977 | 2977 | In-plane CH3 asym. stretch |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.M.; Lee, Y.R.; Kwon, C.H. Conformational Structures of Neutral and Cationic Pivaldehyde Revealed by IR-Resonant VUV-MATI Mass Spectroscopy. Int. J. Mol. Sci. 2022, 23, 14777. https://doi.org/10.3390/ijms232314777
Park SM, Lee YR, Kwon CH. Conformational Structures of Neutral and Cationic Pivaldehyde Revealed by IR-Resonant VUV-MATI Mass Spectroscopy. International Journal of Molecular Sciences. 2022; 23(23):14777. https://doi.org/10.3390/ijms232314777
Chicago/Turabian StylePark, Sung Man, Yu Ran Lee, and Chan Ho Kwon. 2022. "Conformational Structures of Neutral and Cationic Pivaldehyde Revealed by IR-Resonant VUV-MATI Mass Spectroscopy" International Journal of Molecular Sciences 23, no. 23: 14777. https://doi.org/10.3390/ijms232314777