
Design and Validation of a Sensitive Multisteroid LC-MS/MS Assay for the Routine Clinical Use: One-Step Sample Preparation with Phospholipid Removal and Comparison to Immunoassays


Abstract
:1. Introduction
2. Results
2.1. Method Development and Validation
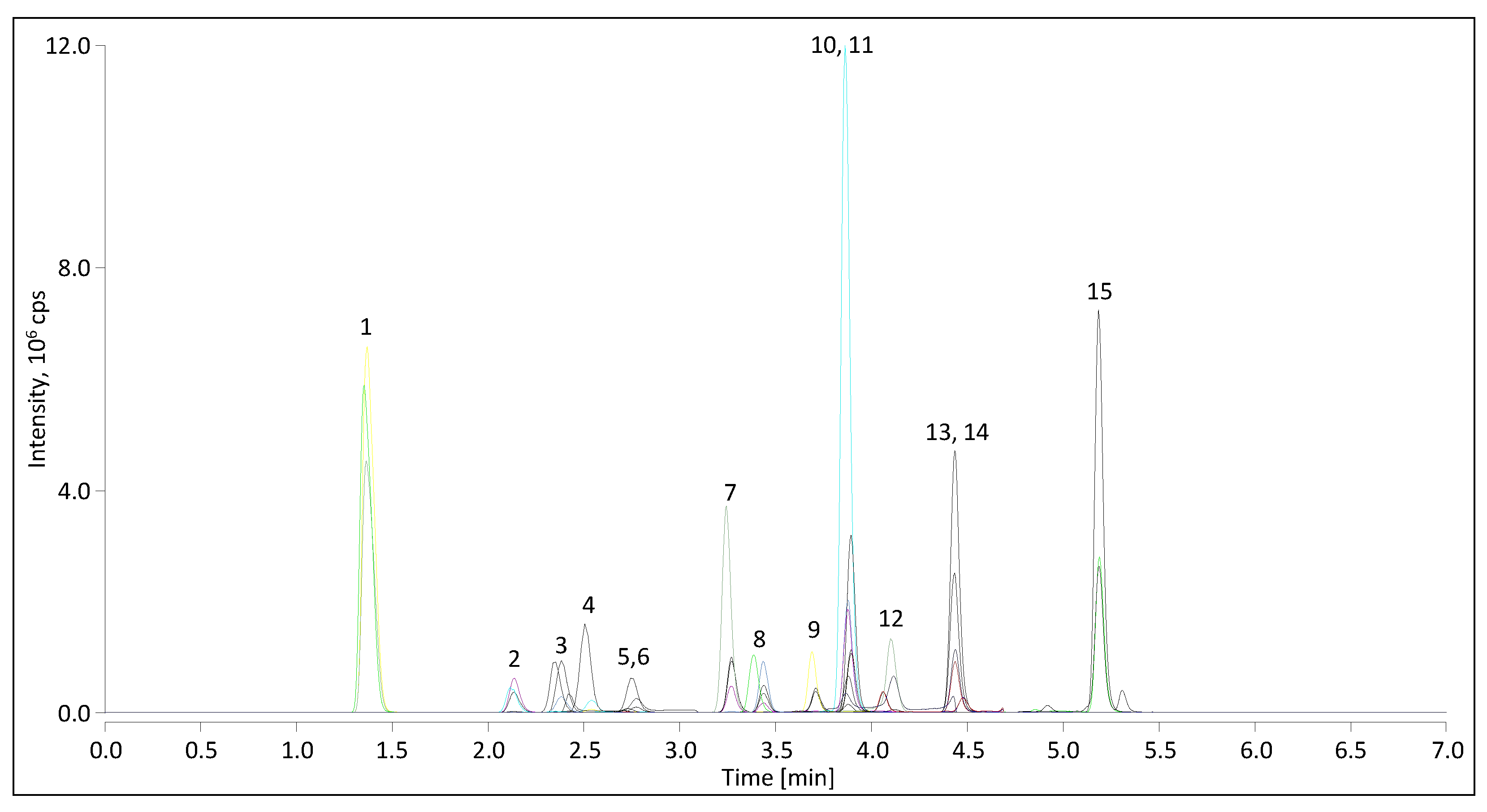
2.1.1. Mass Spectrometry
2.1.2. Liquid Chromatography
2.1.3. Sample Preparation
2.2. Validation
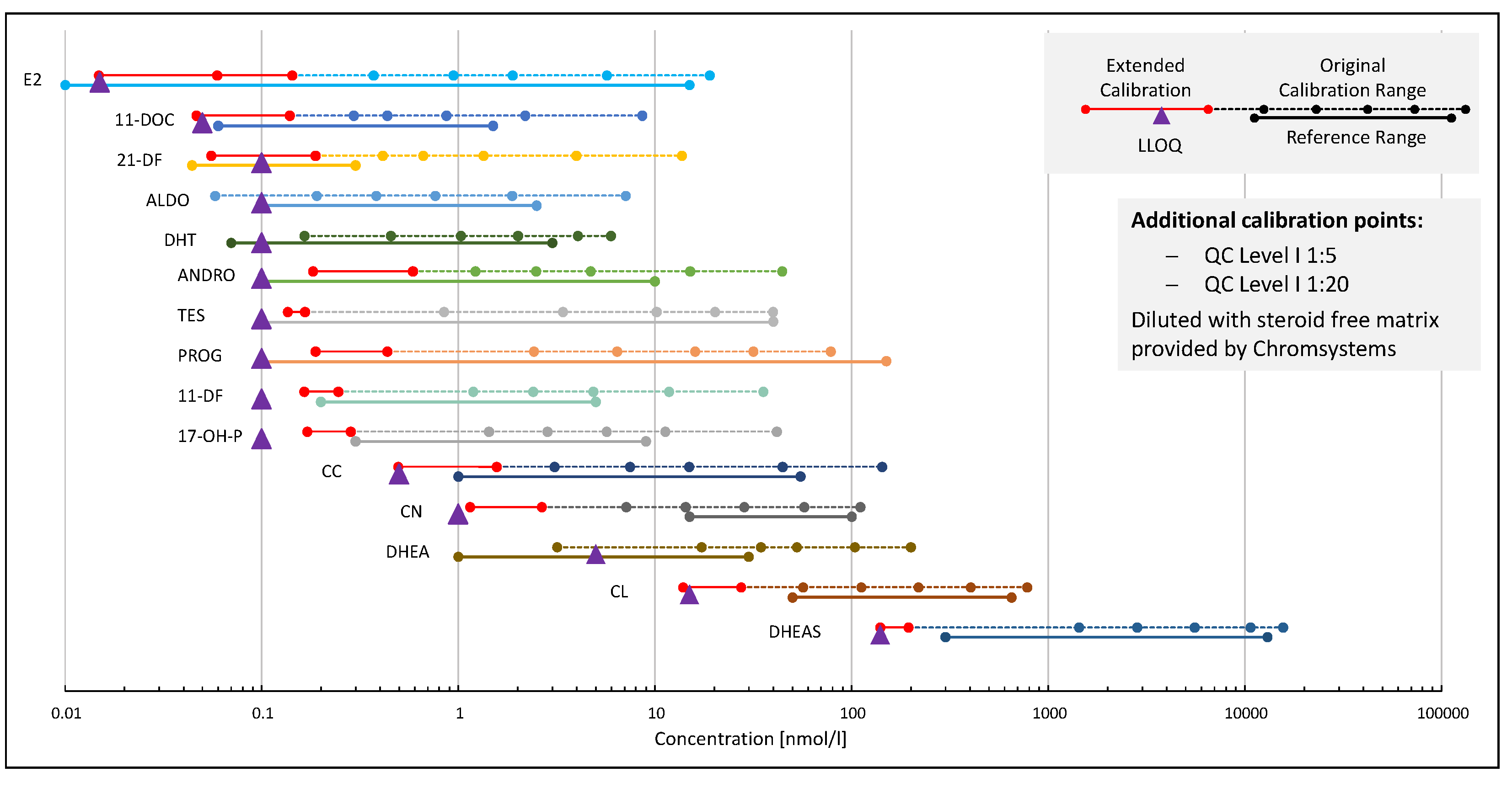
2.2.1. Calibration, Regression Model and Linearity
2.2.2. Sensitivity and Limits of Quantification
2.2.3. Imprecision
2.2.4. Accuracy
2.2.5. Selectivity and Specificity
2.2.6. Interferences
2.2.7. Stability and Robustness
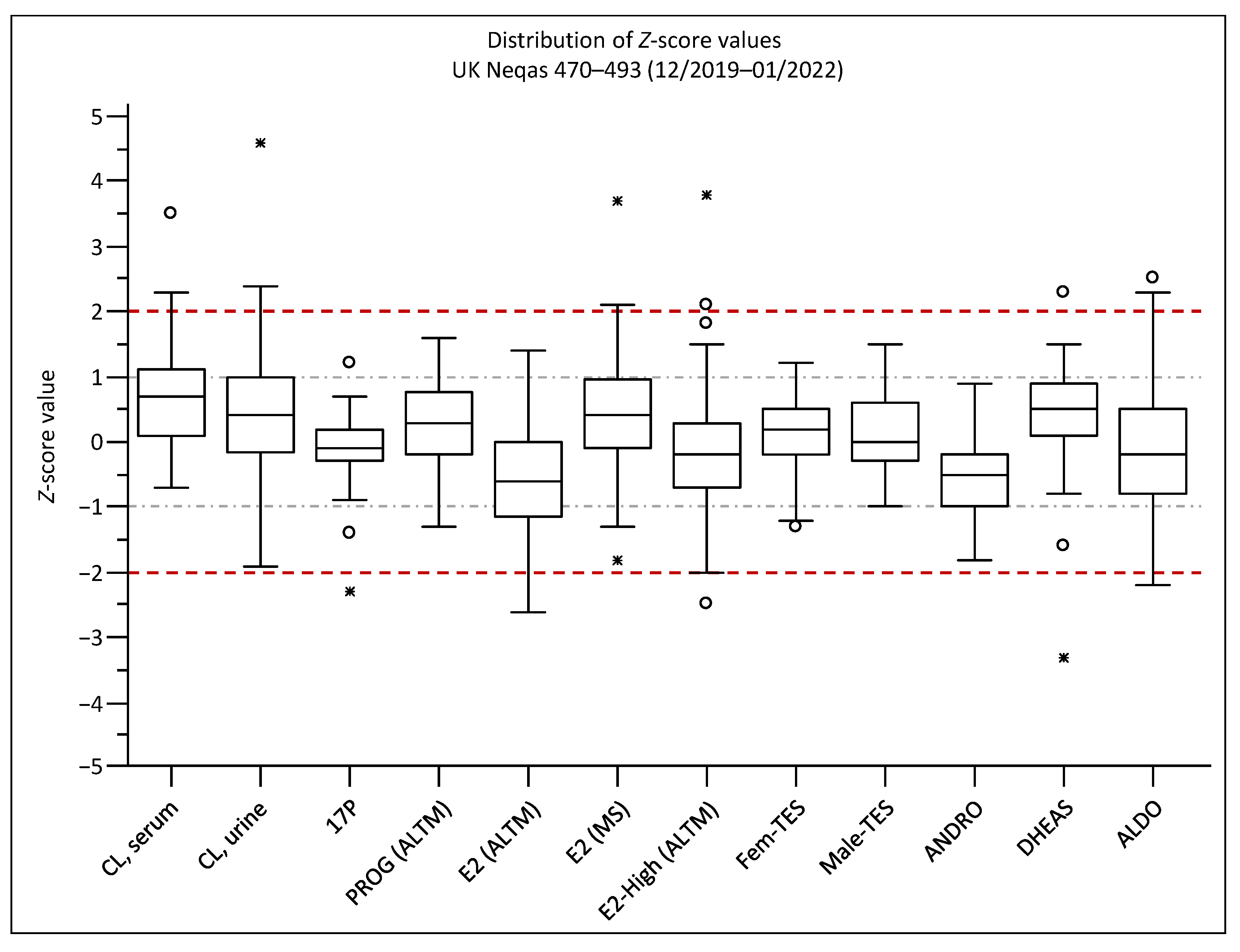
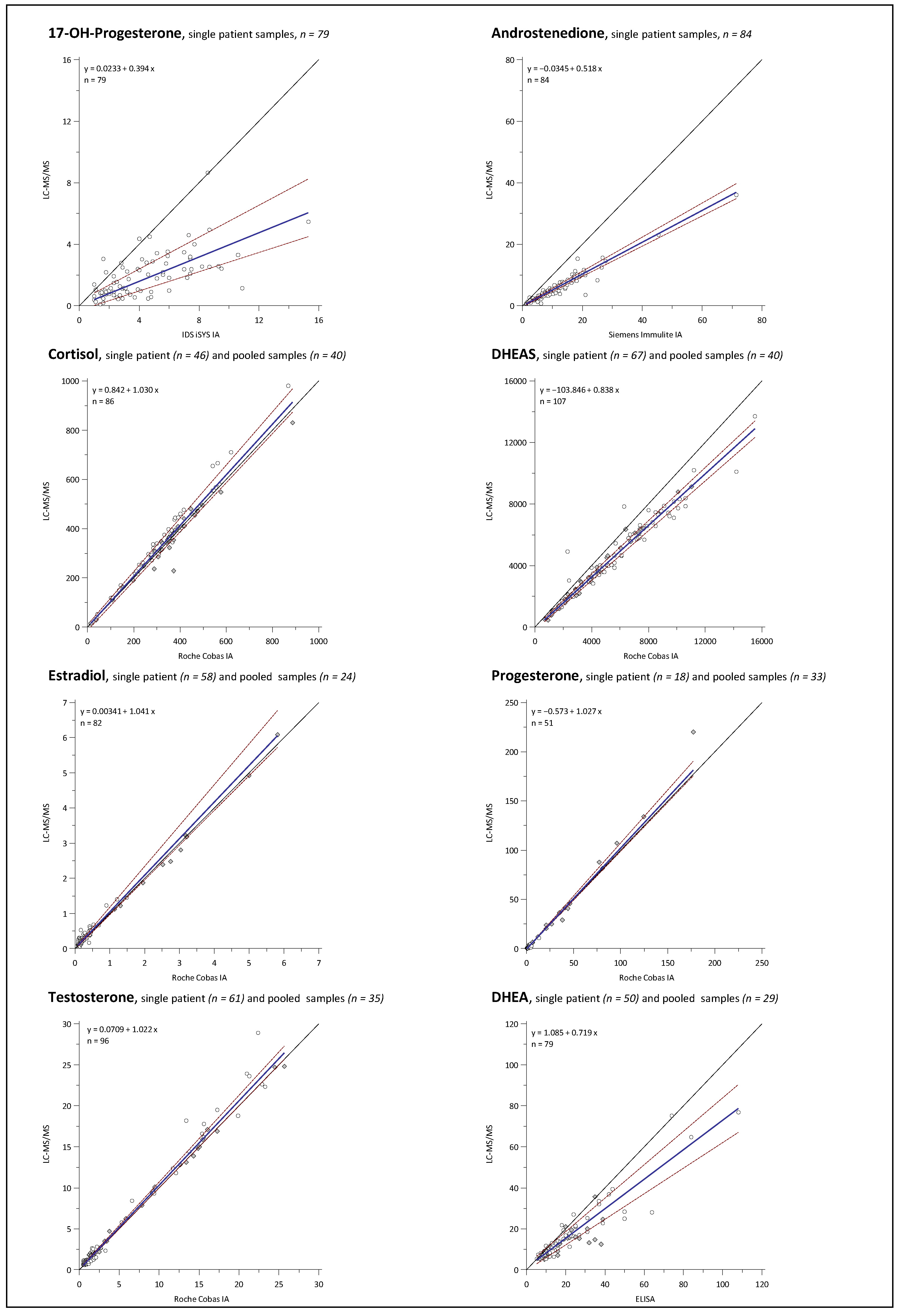
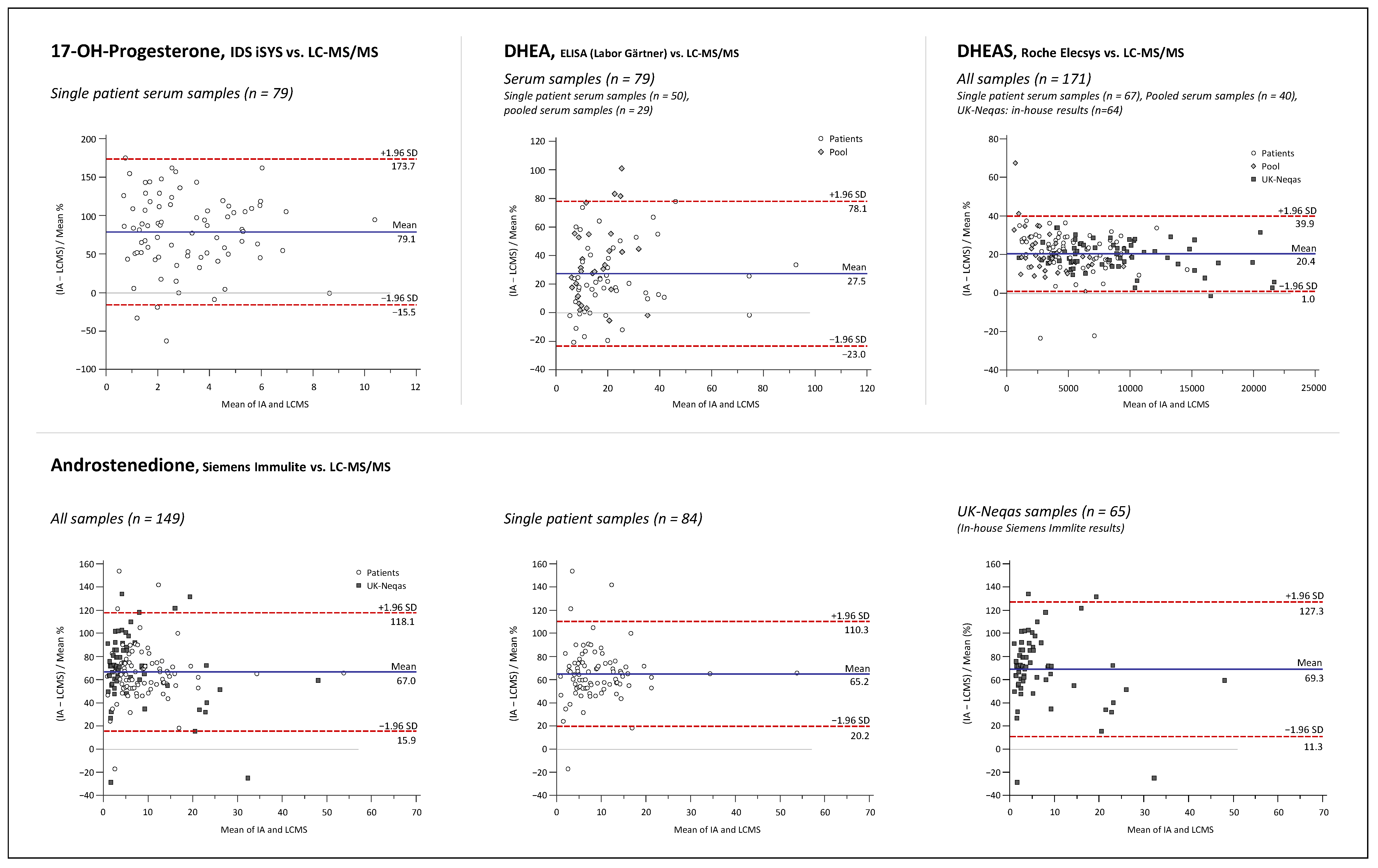
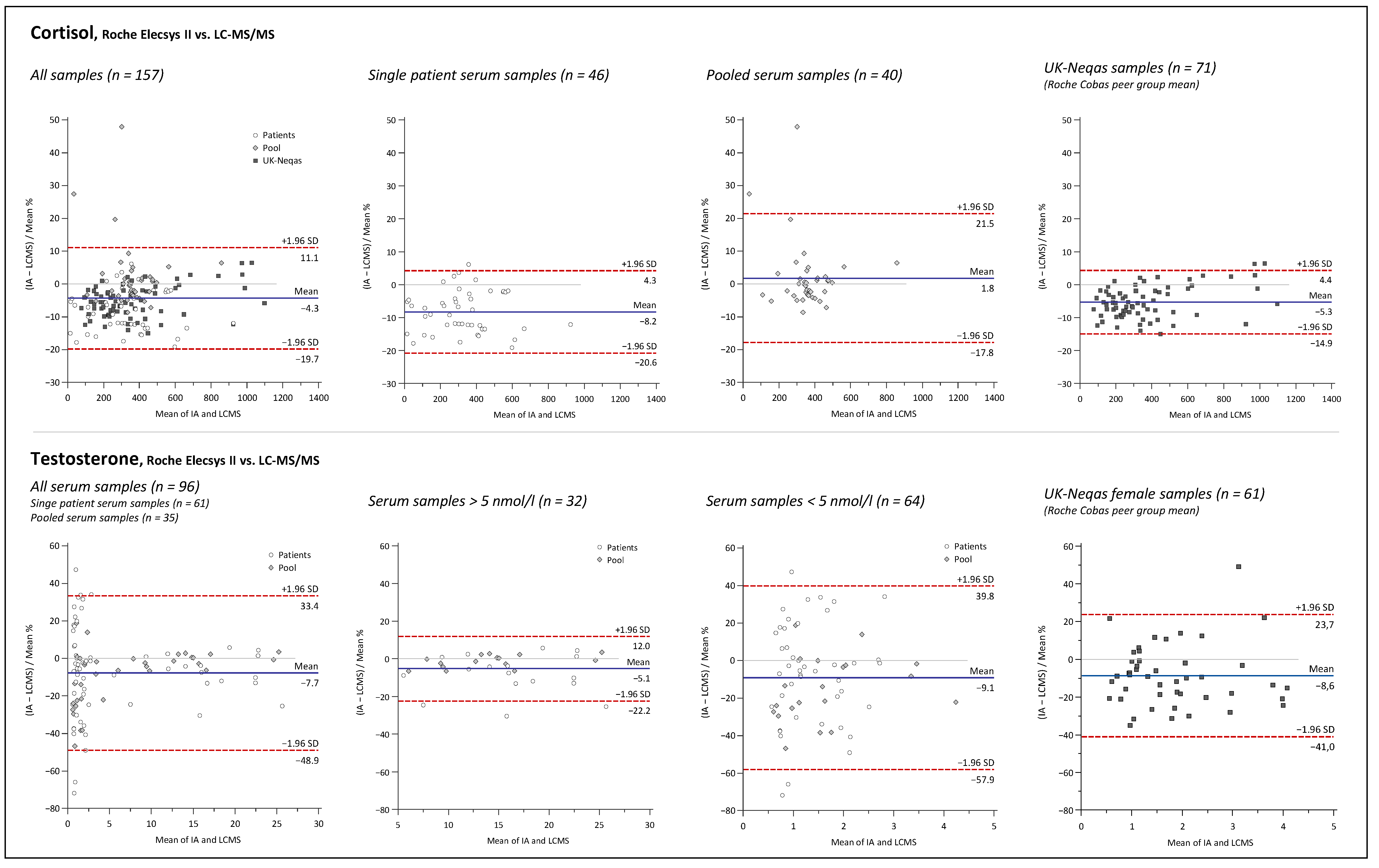
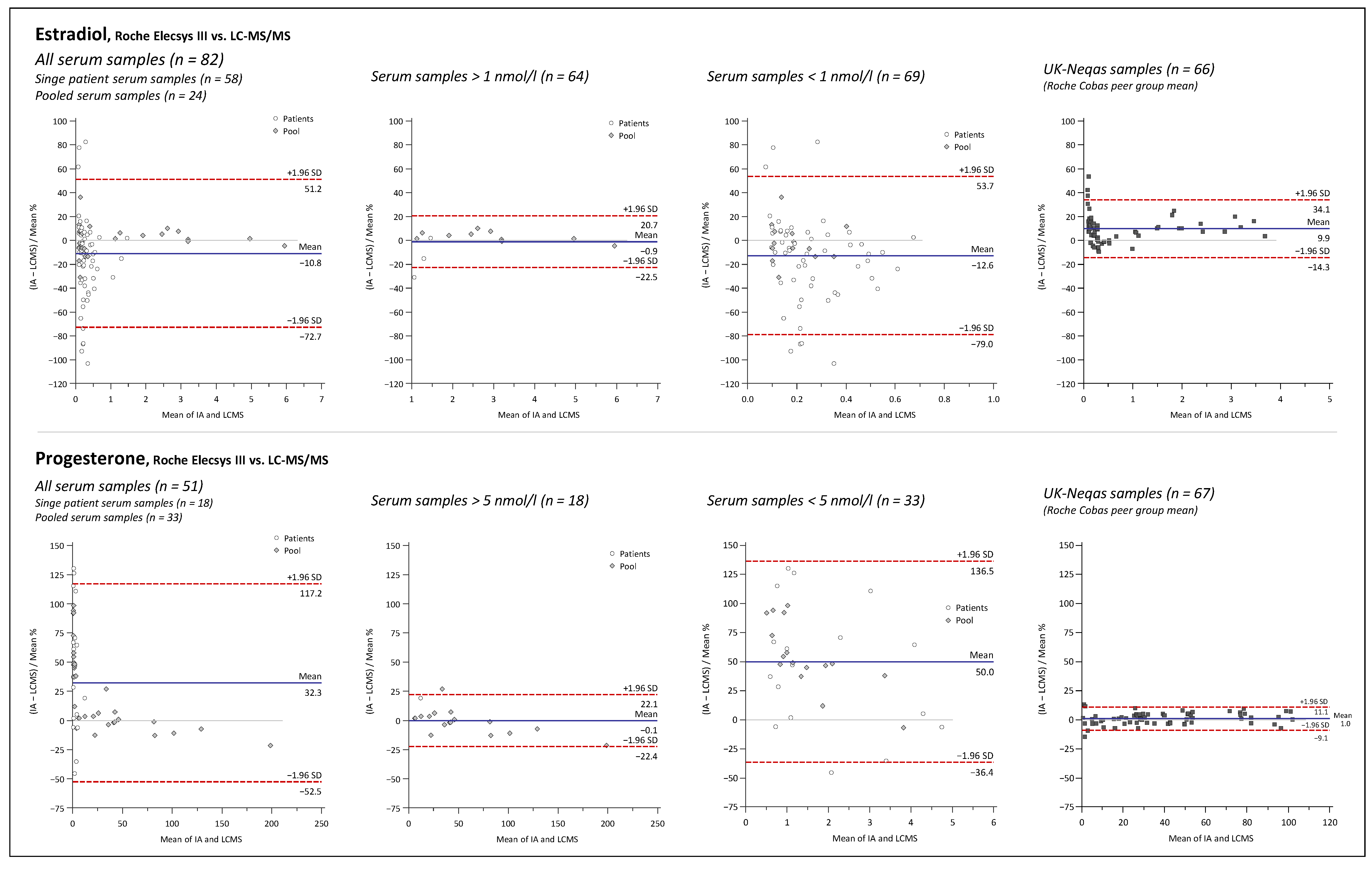
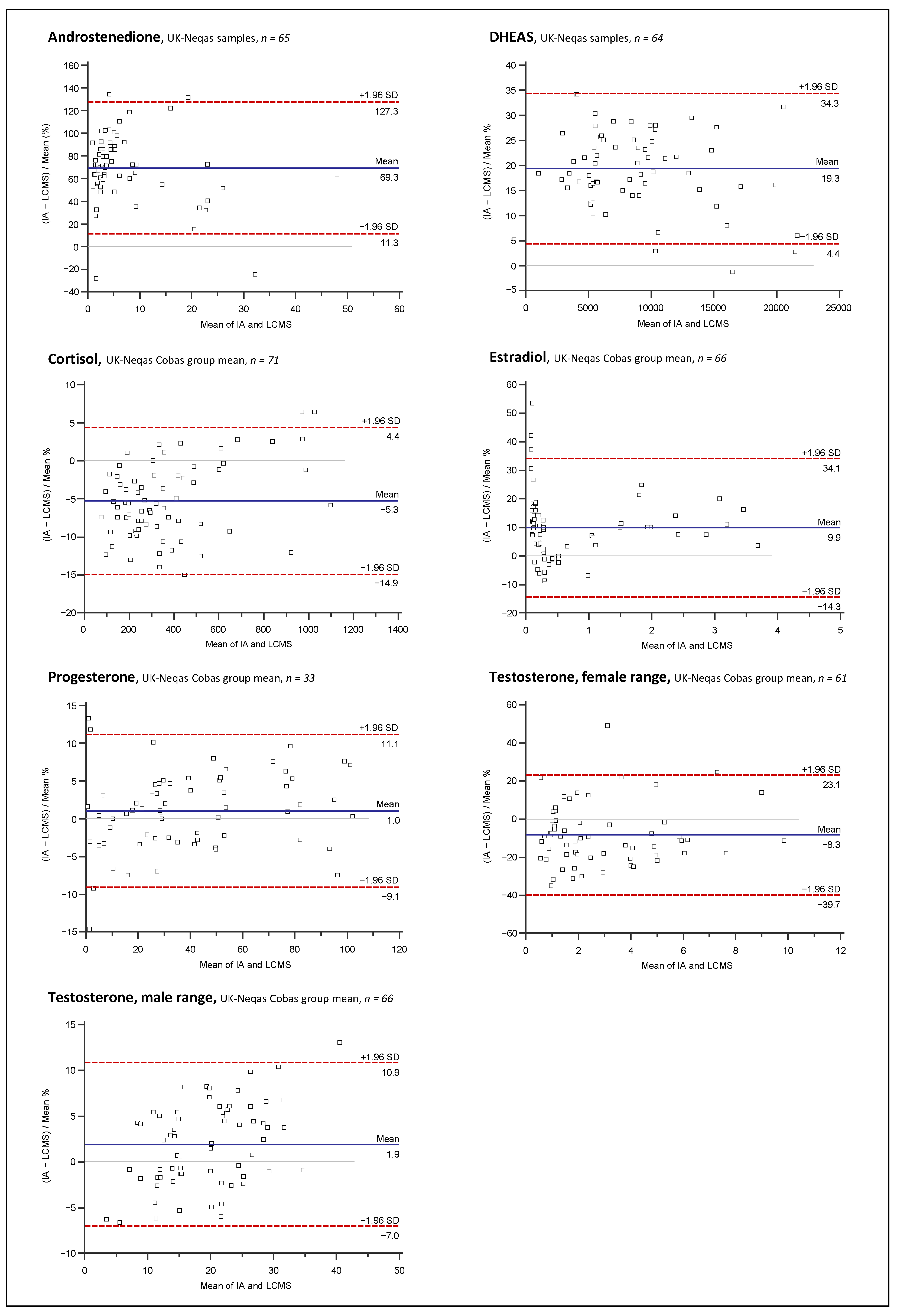
2.3. Assay Comparison
3. Discussion
3.1. Method Development
3.2. Assay Validation
3.3. Assay Comparison
4. Materials and Methods
4.1. Substances and Consumables
4.2. Preparation of Reagents and Mobile Phases
4.3. Calibrators, Controls, and Serum Samples
4.4. Sample Preparation
4.5. Liquid Chromatography–Mass Spectrometry
4.6. Optimizing Mass Spectrometer Settings
4.7. Data Recording and Processing
4.8. Method Validation
4.8.1. Linearity and Calibration Model
4.8.2. Sensitivity
4.8.3. Precision and Accuracy
4.8.4. Proficiency Testing
4.8.5. Recovery
4.8.6. Selectivity and Specificity—Postcolumn Infusion Experiments
4.8.7. Matrix Effects and Extraction Recovery
4.8.8. Interferences
4.8.9. Stability
4.9. Assay Comparison
4.9.1. Immunoassays
4.9.2. Single-Patient Samples
4.9.3. Pooled Serum Samples
4.9.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Honour, J.W. Diagnosis of Diseases of Steroid Hormone Production, Metabolism and Action. J. Clin. Res. Pediatr. Endocrinol. 2009, 1, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.E.; Keevil, B.; Huhtaniemi, I.T. Mass Spectrometry and Immunoassay: How to Measure Steroid Hormones Today and Tomorrow. Eur. J. Endocrinol. 2015, 173, D1–D12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesti, E.; Boccard, J.; Visconti, G.; González-Ruiz, V.; Rudaz, S. From a Single Steroid to the Steroidome: Trends and Analytical Challenges. J. Steroid Biochem. Mol. Biol. 2021, 206, 105797. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J. Matrix Effects: The Achilles Heel of Quantitative High-Performance Liquid Chromatography-Electrospray-Tandem Mass Spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef]
- Ma, Y.-C.; Kim, H.-Y. Determination of Steroids by Liquid Chromatography/Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1997, 8, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Häkkinen, M.R.; Heinosalo, T.; Saarinen, N.; Linnanen, T.; Voutilainen, R.; Lakka, T.; Jääskeläinen, J.; Poutanen, M.; Auriola, S. Analysis by LC–MS/MS of Endogenous Steroids from Human Serum, Plasma, Endometrium and Endometriotic Tissue. J. Pharm. Biomed. Anal. 2018, 152, 165–172. [Google Scholar] [CrossRef]
- Gaudl, A.; Kratzsch, J.; Ceglarek, U. Advancement in Steroid Hormone Analysis by LC–MS/MS in Clinical Routine Diagnostics—A Three Year Recap from Serum Cortisol to Dried Blood 17α-Hydroxyprogesterone. J. Steroid Biochem. Mol. Biol. 2019, 192, 105389. [Google Scholar] [CrossRef]
- Fiers, T.; Casetta, B.; Bernaert, B.; Vandersypt, E.; Debock, M.; Kaufman, J.-M. Development of a Highly Sensitive Method for the Quantification of Estrone and Estradiol in Serum by Liquid Chromatography Tandem Mass Spectrometry without Derivatization. J. Chromatogr. B 2012, 893–894, 57–62. [Google Scholar] [CrossRef]
- Lindner, J.M.; Vogeser, M.; Grimm, S.H. Biphenyl Based Stationary Phases for Improved Selectivity in Complex Steroid Assays. J. Pharm. Biomed. Anal. 2017, 142, 66–73. [Google Scholar] [CrossRef]
- Takkis, K.; Aro, R.; Kõrgvee, L.T.; Varendi, H.; Lass, J.; Herodes, K.; Kipper, K. Signal Enhancement in the HPLC-ESI-MS/MS Analysis of Spironolactone and Its Metabolites Using HFIP and NH4F as Eluent Additives. Anal. Bioanal. Chem. 2017, 409, 3145–3151. [Google Scholar] [CrossRef]
- Wang, W.; Cole, R.B. Enhanced Collision-Induced Decomposition Efficiency and Unraveling of Fragmentation Pathways for Anionic Adducts of Brevetoxins in Negative Ion Electrospray Mass Spectrometry. Anal. Chem. 2009, 81, 8826–8838. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. High-Resolution Mass Spectrometry-Based Multi-Residue Method Covering Relevant Steroids, Stilbenes and Resorcylic Acid Lactones in a Variety of Animal-Based Matrices. Anal. Chim. Acta 2019, 1054, 59–73. [Google Scholar] [CrossRef]
- Pesek, J.J.; Matyska, M.T. Ammonium Fluoride as a Mobile Phase Additive in Aqueous Normal Phase Chromatography. J. Chromatogr. A 2015, 1401, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, S.; Antonio, L.; Jans, I.; Lintermans, A.; Neven, P.; Claessens, F.; Decallonne, B.; Billen, J.; Vanderschueren, D.; Vermeersch, P. Sensitive Routine Liquid Chromatography–Tandem Mass Spectrometry Method for Serum Estradiol and Estrone without Derivatization. Anal. Bioanal. Chem. 2013, 405, 8569–8577. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, S.; Jans, I.; Peersman, N.; Billen, J.; Vanderschueren, D.; Desmet, K.; Vermeersch, P. Possibilities and Limitations of Signal Summing for an Immunosuppressant LC-MS/MS Method. Anal. Bioanal. Chem. 2015, 407, 6191–6199. [Google Scholar] [CrossRef]
- Aurand, C.; Trinh, A. Selective Depletion of Phospholipids in Bioanalysis Using HybridSPE-PPT Technology. Chromatography Today, 15 October 2009; 38–41. Available online: https://www.chromatographytoday.com/article/solid-phase-extraction-spe/34/supelco/selective-depletion-of-phospholipids-in-bioanalysis-using-hybridspe-ppt-technology/510(accessed on 6 November 2022).
- Ahmad, S.; Kalra, H.; Gupta, A.; Raut, B.; Hussain, A.; Rahman, M.A. HybridSPE: A Novel Technique to Reduce Phospholipid-Based Matrix Effect in LC-ESI-MS Bioanalysis. J. Pharm. Bioallied Sci. 2012, 4, 267–275. [Google Scholar] [CrossRef]
- Aurand, C. Sample Prep Method Development and Optimization for the LC/MS/MS Analysis of Steroid Hormones in Plasma; Reporter 32.2. 2015, pp. 12–14. Available online: https://theanalyticalscientist.com/fileadmin/tas/issues/App_Notes/00515-app-note-sigma-supplied.pdf (accessed on 6 November 2022).
- Kim, S.Y.; Kwon, N.H.; Cheong, J.C.; Kim, J.Y. LC–MS/MS Method for Determining Picogram-Level of Zolpidem and Its Main Metabolites in Hair Using a Zirconia-Based Sorbent. Talanta 2021, 228, 122041. [Google Scholar] [CrossRef]
- Blackwell, J.A.; Carr, P.W. Development of an Eluotropic Series for the Chromatography of Lewis Bases on Zirconium Oxide. Anal. Chem. 1992, 64, 863–873. [Google Scholar] [CrossRef]
- Desharnais, B.; Camirand-Lemyre, F.; Mireault, P.; Skinner, C.D. Procedure for the Selection and Validation of a Calibration Model I—Description and Application. J. Anal. Toxicol. 2017, 41, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Desharnais, B.; Camirand-Lemyre, F.; Mireault, P.; Skinner, C.D. Procedure for the Selection and Validation of a Calibration Model II—Theoretical Basis. J. Anal. Toxicol. 2017, 41, 269–276. [Google Scholar] [CrossRef]
- Almeida, A.M.; Castel-Branco, M.M.; Falcão, A.C. Linear Regression for Calibration Lines Revisited: Weighting Schemes for Bioanalytical Methods. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 774, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Peitzsch, M.; Kaden, D.; Langton, K.; Pamporaki, C.; Masjkur, J.; Tsatsaronis, G.; Mangelis, A.; Williams, T.A.; Reincke, M.; et al. Reference Intervals for Plasma Concentrations of Adrenal Steroids Measured by LC-MS/MS: Impact of Gender, Age, Oral Contraceptives, Body Mass Index and Blood Pressure Status. Clin. Chim. Acta 2017, 470, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Mezzullo, M.; Pelusi, C.; Fazzini, A.; Repaci, A.; Di Dalmazi, G.; Gambineri, A.; Pagotto, U.; Fanelli, F. Female and Male Serum Reference Intervals for Challenging Sex and Precursor Steroids by Liquid Chromatography—Tandem Mass Spectrometry. J. Steroid Biochem. Mol. Biol. 2020, 197, 105538. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, R.; King, R.C.; Olah, T.V.; Merkle, K. The Effects of Sample Preparation Methods on the Variability of the Electrospray Ionization Response for Model Drug Compounds. Rapid Commun. Mass Spectrom. 1999, 13, 1175–1185. [Google Scholar] [CrossRef]
- Neville, D.; Houghton, R.; Garrett, S. Efficacy of Plasma Phospholipid Removal during Sample Preparation and Subsequent Retention under Typical UHPLC Conditions. Bioanalysis 2012, 4, 795–807. [Google Scholar] [CrossRef]
- Desai, R.; Harwood, D.T.; Handelsman, D.J. Simultaneous Measurement of 18 Steroids in Human and Mouse Serum by Liquid Chromatography–Mass Spectrometry without Derivatization to Profile the Classical and Alternate Pathways of Androgen Synthesis and Metabolism. Clin. Mass Spectrom. 2019, 11, 42–51. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- Bienvenu, J.F.; Provencher, G.; Bélanger, P.; Bérubé, R.; Dumas, P.; Gagné, S.; Gaudreau, É.; Fleury, N. Standardized Procedure for the Simultaneous Determination of the Matrix Effect, Recovery, Process Efficiency, and Internal Standard Association. Anal. Chem. 2017, 89, 7560–7568. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, L.; Wang, J.; Lin, H.; Ke, P.; Zhuang, J.; Huang, X. Simultaneous Quantitation of Endogenous Estrone, 17Β-Estradiol, and Estriol in Human Serum by Isotope-Dilution Liquid Chromatography–Tandem Mass Spectrometry for Clinical Laboratory Applications. Anal. Bioanal. Chem. 2017, 409, 2627–2638. [Google Scholar] [CrossRef]
- Kushnir, M.M.; Rockwood, A.L.; Bergquist, J.; Varshavsky, M.; Roberts, W.L.; Yue, B.; Bunker, A.M.; Meikle, A.W. High-Sensitivity Tandem Mass Spectrometry Assay for Serum Estrone and Estradiol. Am. J. Clin. Pathol. 2008, 129, 530–539. [Google Scholar] [CrossRef]
- Botelho, J.C.; Ribera, A.; Cooper, H.C.; Vesper, H.W. Evaluation of an Isotope Dilution HPLC Tandem Mass Spectrometry Candidate Reference Measurement Procedure for Total 17-β Estradiol in Human Serum. Anal. Chem. 2016, 88, 11123–11129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, B.-E.E.; Kellmann, R.; Viste, K.; Bjørnevik, A.T.; Eikesdal, H.P.; Lønning, P.E.; Sagen, J.V.; Almås, B. An Ultrasensitive Routine LC-MS/MS Method for Estradiol and Estrone in the Clinically Relevant Sub-Picomolar Range. J. Endocr. Soc. 2020, 4, bvaa047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courant, F.; Antignac, J.-P.; Maume, D.; Monteau, F.; Andersson, A.-M.; Skakkebaek, N.; Andre, F.; Le Bizec, B. Exposure Assessment of Prepubertal Children to Steroid Endocrine Disrupters. Anal. Chim. Acta 2007, 586, 105–114. [Google Scholar] [CrossRef]
- Hobe, G.; Schön, R.; Goncharov, N.; Katsiya, G.; Koryakin, M.; Gesson-Cholat, I.; Oettel, M.; Zimmermann, H. Some New Aspects of 17α-Estradiol Metabolism in Man. Steroids 2002, 67, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Li, L.; Gatson, J.W.; Maass, D.; Wigginton, J.G.; Simpkins, J.W.; Schug, K.A. Simultaneous Quantification of Four Native Estrogen Hormones at Trace Levels in Human Cerebrospinal Fluid Using Liquid Chromatography-Tandem Mass Spectrometry. J. Pharm. Biomed. Anal. 2011, 54, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Beinhauer, J.; Bian, L.; Fan, H.; Šebela, M.; Kukula, M.; Barrera, J.A.; Schug, K.A. Bulk Derivatization and Cation Exchange Restricted Access Media-Based Trap-and-Elute Liquid Chromatography-Mass Spectrometry Method for Determination of Trace Estrogens in Serum. Anal. Chim. Acta 2015, 858, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Lee, S.Y.; Lee, H.-J.; Yoon, N.-Y.; Lee, W.S. The Efficacy and Safety of 17a-Estradiol (Ell-Cranell® Alpha 0.025%) Solution on Female Pattern Hair Loss: Single Center, Open-Label, Non-Comparative, Phase IV Study. Ann. Dermatol. 2012, 24, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Blume-Peytavi, U.; Kunte, C.; Krisp, A.; Bartels, N.G.; Ellwanger, U.; Hoffmann, R. Comparison of the Efficacy and Safety of Topical Minoxidil and Topical Alfatradiol in the Treatment of Androgenetic Alopecia in Women. JDDG J. Ger. Soc. Dermatol. 2007, 5, 391–395. [Google Scholar] [CrossRef]
- Hawley, J.M.; Owen, L.J.; Debono, M.; Newell-Price, J.; Keevil, B.G. Development of a Rapid Liquid Chromatography Tandem Mass Spectrometry Method for the Quantitation of Serum Dexamethasone and Its Clinical Verification. Ann. Clin. Biochem. 2018, 55, 665–672. [Google Scholar] [CrossRef]
- Mcwhinney, B.C.; Briscoe, S.E.; Ungerer, J.P.J.; Pretorius, C.J. Measurement of Cortisol, Cortisone, Prednisolone, Dexamethasone and 11-Deoxycortisol with Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry: Application for Plasma, Plasma Ultrafiltrate, Urine and Saliva in a Routine Laboratory. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 2863–2869. [Google Scholar] [CrossRef]
- Antignac, J.P.; Le Bizec, B.; Monteau, F.; Poulain, F.; André, F. Collision-Induced Dissociation of Corticosteroids in Electrospray Tandem Mass Spectrometry and Development of a Screening Method by High Performance Liquid Chromatography/Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 33–39. [Google Scholar] [CrossRef]
- Ionita, I.A.; Fast, D.M.; Akhlaghi, F. Development of a Sensitive and Selective Method for the Quantitative Analysis of Cortisol, Cortisone, Prednisolone and Prednisone in Human Plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 765–772. [Google Scholar] [CrossRef]
- Van Der Gugten, J.G.; Dubland, J.; Liu, H.F.; Wang, A.; Joseph, C.; Holmes, D.T. Determination of Serum Aldosterone by Liquid Chromatography and Tandem Mass Spectrometry: A Liquid-Liquid Extraction Method for the ABSCIEX API-5000 Mass Spectrometry System. J. Clin. Pathol. 2012, 65, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Passing, H.; Bablok, W. A New Biometrical Procedure for Testing the Equality of Measurements from Two Different Analytical Methods. Application of Linear Regression Procedures for Method Comparison Studies in Clinical Chemistry, Part I. Clin. Chem. Lab. Med. 1983, 21, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin Bland, J.; Altman, D. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1986, 327, 307–310. [Google Scholar] [CrossRef]
- Bablok, W.; Passing, H. Application of Statistical Procedures in Analytical Instrument Testing. J. Automat. Chem. 1985, 7, 74–79. [Google Scholar] [CrossRef]
- Bilic-Zulle, L. Comparison of Methods: Passing and Bablok Regression. Biochem. Med. 2011, 21, 49–52. [Google Scholar] [CrossRef]
- Rousu, T.; Tolonen, A. Comparison of Unit Resolution SRM and TOF-MS at 12,000 Mass Resolution for Quantitative Bioanalysis of 11 Steroids from Human Plasma. Bioanalysis 2012, 4, 555–563. [Google Scholar] [CrossRef]
- Maeda, N.; Tanaka, E.; Suzuki, T.; Okumura, K.; Nomura, S.; Miyasho, T.; Haeno, S.; Yokota, H. Accurate Determination of Tissue Steroid Hormones, Precursors and Conjugates in Adult Male Rat. J. Biochem. 2013, 153, 63–71. [Google Scholar] [CrossRef]
- Woźniak, B.; Matraszek-Zuchowska, I.; Witek, S.; Posyniak, A. Development of LC-MS/MS Confirmatory Method for the Determination of Testosterone in Bovine Serum. J. Vet. Res. 2017, 61, 81–89. [Google Scholar] [CrossRef]
- Lih, F.B.; Titus, M.A.; Mohler, J.L.; Tomer, K.B. Atmospheric Pressure Photoionization Tandem Mass Spectrometry of Androgens in Prostate Cancer. Anal. Chem. 2010, 82, 6000–6007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, A.J.; Ducroq, D.H.; Neale, S.M.; Wise, M.P.; Mitchem, K.L.; Armston, A.; Barth, J.H.; El-Farhan, N.; Rees, D.A.; Evans, C. The Effect of Serum Matrix and Gender on Cortisol Measurement by Commonly Used Immunoassays. Ann. Clin. Biochem. 2014, 51, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, J.M.; Owen, L.J.; Lockhart, S.J.; Monaghan, P.J.; Armston, A.; Chadwick, C.A.; Wilshaw, H.; Freire, M.; Perry, L.; Keevil, B.G. Serum Cortisol: An up-to-Date Assessment of Routine Assay Performance. Clin. Chem. 2016, 62, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanelli, F.; Belluomo, I.; Di Lallo, V.D.; Cuomo, G.; De Iasio, R.; Baccini, M.; Casadio, E.; Casetta, B.; Vicennati, V.; Gambineri, A.; et al. Serum Steroid Profiling by Isotopic Dilution-Liquid Chromatography-Mass Spectrometry: Comparison with Current Immunoassays and Reference Intervals in Healthy Adults. Steroids 2011, 76, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttler, R.M.; Kruit, A.; Blankenstein, M.A.; Heijboer, A.C. Measurement of Dehydroepiandrosterone Sulphate (DHEAS): A Comparison of Isotope-Dilution Liquid Chromatography Tandem Mass Spectrometry (Id-Lc-Ms/Ms) and Seven Currently Available Immunoassays. Clin. Chim. Acta 2013, 424, 22–26. [Google Scholar] [CrossRef]
- La’ulu, S.L.; Kalp, K.J.; Straseski, J.A. How Low Can You Go? Analytical Performance of Five Automated Testosterone Immunoassays. Clin. Biochem. 2018, 58, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Bowers, K.; Kroner, G.; Payto, D.; Colón-Franco, J. Comparison of Three Serum Androstenedione Assays: The Siemens Immulite Immunoassay, the Roche Elecsys Immunoassay, and an LC-MS/MS Assay. Am. J. Clin. Pathol. 2021, 156, S10–S11. [Google Scholar] [CrossRef]
- Koal, T.; Schmiederer, D.; Pham-Tuan, H.; Röhring, C.; Rauh, M. Standardized LC-MS/MS Based Steroid Hormone Profile-Analysis. J. Steroid Biochem. Mol. Biol. 2012, 129, 129–138. [Google Scholar] [CrossRef]
- Roli, L.; Santi, D.; Belli, S.; Tagliavini, S.; Cavalieri, S.; De Santis, M.C.; Baraldi, E.; Fanelli, F.; Mezzullo, M.; Granata, A.R.; et al. The Steroid Response to Human Chorionic Gonadotropin (HCG) Stimulation in Men with Klinefelter Syndrome Does Not Change Using Immunoassay or Mass Spectrometry. J. Endocrinol. Investig. 2017, 40, 841–850. [Google Scholar] [CrossRef]
- Van der Veen, A.; van Faassen, M.; de Jong, W.H.A.; van Beek, A.P.; Dijck-Brouwer, D.A.J.; Kema, I.P. Development and Validation of a LC-MS/MS Method for the Establishment of Reference Intervals and Biological Variation for Five Plasma Steroid Hormones. Clin. Biochem. 2019, 68, 15–23. [Google Scholar] [CrossRef]
- Olisov, D.; Lee, K.; Jun, S.H.; Song, S.H.; Kim, J.H.; Lee, Y.A.; Shin, C.H.; Song, J. Measurement of Serum Steroid Profiles by HPLC-Tandem Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1117, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gervasoni, J.; Schiattarella, A.; Primiano, A.; D’Addurno, I.; Cocci, A.; Zuppi, C.; Persichilli, S. Simultaneous Quantification of 17-Hydroxyprogesterone, Androstenedione, Testosterone and Cortisol in Human Serum by LC-MS/MS Using TurboFlow Online Sample Extraction. Clin. Biochem. 2016, 49, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Rauh, M.; Gröschl, M.; Rascher, W.; Dörr, H.G. Automated, Fast and Sensitive Quantification of 17α-Hydroxy-Progesterone, Androstenedione and Testosterone by Tandem Mass Spectrometry with on-Line Extraction. Steroids 2006, 71, 450–458. [Google Scholar] [CrossRef] [PubMed]
- UK-Neqas Steroid Hormones Scheme Discription. Available online: https://birminghamquality.org.uk/eqa-programmes/ster/ (accessed on 6 November 2022).
- UK-Neqas Birmingham Quality Participants Manual. Available online: https://birminghamquality.org.uk/assets/in-house/doc/man/M-MAN-001ParticipantsManualV9.0.pdf (accessed on 6 November 2022).
- Grant, R.P.; Rappold, B.A. Development and Validation of Small Molecule Analytes by Liquid Chromatography-Tandem Mass Spectrometry. In Principles and Applications of Clinical Mass Spectrometry, 1st ed.; Rifai, N., Horvath, R.A., Wittwer, C.T., Hoofnagle, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 115–179. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Parameter | Unit | QC-I 1:20 | QC-I 1:10 | QC-I 1:5 | QC-I | QC-II | QC-III |
|---|---|---|---|---|---|---|---|---|
| 11-DOC | (nmol/L) | 0.012 | 0.023 | 0.047 | 0.233 | 0.590 | 3.050 | |
| IM-IN | (%) | n.a. | 13.5 | 9.0 | 4.7 | 3.3 | 2.7 | |
| IM-ALL | (%) | n.a. | 6.3 | 5.7 | 1.9 | 1.2 | 0.9 | |
| BI-ALL | (%) | n.a. | 14.5 | 6.9 | 1.4 | −1.8 | −1.3 | |
| 17P | (nmol/L) | 0.04 | 0.09 | 0.17 | 0.85 | 4.30 | 25.80 | |
| IM-IN | (%) | 14.3 | 7.3 | 5.3 | 3.4 | 2.1 | 1.2 | |
| IM-ALL | (%) | 7.9 | 6.1 | 3.9 | 1.4 | 0.8 | 0.6 | |
| BI-ALL | (%) | 1.9 | 0.3 | 0.6 | −0.5 | −0.3 | −1.6 | |
| ANDRO | (nmol/L) | 0.05 | 0.09 | 0.18 | 0.91 | 3.75 | 31.90 | |
| IM-IN | (%) | 11.2 | 6.0 | 5.5 | 2.6 | 2.0 | 2.5 | |
| IM-ALL | (%) | 6.1 | 4.5 | 3.6 | 1.2 | 0.8 | 0.8 | |
| BI-ALL | (%) | 5.1 | −0.8 | 1.5 | −0.4 | 1.4 | −2.4 | |
| DHEA | (nmol/L) | 0.3 | 0.7 | 1.4 | 6.8 | 42.6 | 144.0 | |
| IM-IN | (%) | n.a. | n.a. | 23.9 | 7.2 | 5.0 | 3.4 | |
| IM-ALL | (%) | n.a. | n.a. | 11.5 | 4.3 | 1.9 | 2.0 | |
| BI-ALL | (%) | n.a. | n.a. | 11.2 | −1.2 | −0.9 | −2.1 | |
| DHT | (nmol/L) | 0.017 | 0.033 | 0.066 | 0.33 | 1.590 | 3.890 | |
| IM-IN | (%) | n.a. | 45.5 | 26.2 | 8.3 | 5.8 | 6.0 | |
| IM-ALL | (%) | n.a. | 14.2 | 10.0 | 6.3 | 2.8 | 3.5 | |
| BI-ALL | (%) | n.a. | 91.3 | 25.4 | −1.1 | 1.4 | −2.3 | |
| PROG | (nmol/L) | 0.05 | 0.09 | 0.19 | 0.94 | 9.72 | 47.90 | |
| IM-IN | (%) | 21.7 | 8.0 | 6.3 | 2.8 | 2.0 | 1.5 | |
| IM-ALL | (%) | 4.5 | 3.8 | 1.8 | 0.8 | 0.4 | 0.3 | |
| BI-ALL | (%) | 20.6 | 7.8 | 2.2 | −2.5 | 1.5 | −0.4 | |
| TES | (nmol/L) | 0.03 | 0.07 | 0.14 | 0.68 | 5.13 | 26.50 | |
| IM-IN | (%) | 18.6 | 11.1 | 5.6 | 3.9 | 2.3 | 2.5 | |
| IM-ALL | (%) | 13.8 | 7.6 | 4.1 | 2.0 | 1.1 | 1.4 | |
| BI-ALL | (%) | −0.8 | 0.1 | 2.3 | 0.2 | 1.9 | 0.0 | |
| 21-DF | (nmol/L) | 0.01 | 0.03 | 0.06 | 0.28 | 1.05 | 6.58 | |
| IM-IN | (%) | n.a. | n.a. | 18.4 | 10.6 | 9.6 | 10.9 | |
| IM-ALL | (%) | n.a. | n.a. | 11.3 | 5.3 | 3.9 | 2.1 | |
| BI-ALL | (%) | n.a. | n.a. | 6.6 | 0.3 | 7.3 | 5.2 | |
| 11-DF | (nmol/L) | 0.04 | 0.08 | 0.17 | 0.82 | 4.05 | 26.5 | |
| IM-IN | (%) | 12.3 | 7.9 | 4.5 | 11.0 | 3.1 | 2.6 | |
| IM-ALL | (%) | 5.7 | 4.8 | 3.0 | 1.8 | 1.2 | 0.9 | |
| BI-ALL | (%) | 26.1 | 7.5 | 0.9 | −3.1 | −6.9 | 2.9 | |
| CC | (nmol/L) | 0.12 | 0.25 | 0.49 | 2.47 | 12.00 | 80.40 | |
| IM-IN | (%) | 15.6 | 9.6 | 5.1 | 3.5 | 4.4 | 3.5 | |
| IM-ALL | (%) | 7.6 | 5.0 | 3.1 | 1.2 | 0.8 | 1.4 | |
| BI-ALL | (%) | −3.7 | 1.7 | 3.4 | 2.4 | 5.9 | −0.3 | |
| DHEAS | (nmol/L) | 35 | 70 | 140 | 699 | 4230 | 13800 | |
| IM-IN | (%) | 5.2 | 3.4 | 2.0 | 2.2 | 2.1 | 1.1 | |
| IM-ALL | (%) | 0.6 | 0.6 | 0.6 | 0.5 | 0.4 | 0.3 | |
| BI-ALL | (%) | 38.6 | 14.9 | 3.8 | −6.6 | −5.7 | −12.3 | |
| E2 | (nmol/L) | 0.015 | 0.030 | 0.059 | 0.297 | 1.510 | 9.260 | |
| IM-IN | (%) | 10.2 | 9.9 | 9.2 | 6.1 | 5.3 | 4.7 | |
| IM-ALL | (%) | 8.8 | 7.0 | 7.9 | 3.7 | 2.6 | 2.7 | |
| BI-ALL | (%) | 2.4 | −2.1 | −0.8 | −0.5 | −1.3 | −2.7 | |
| CL | (nmol/L) | 3 | 7 | 14 | 69 | 166 | 487 | |
| IM-IN | (%) | 22.7 | 10.7 | 4.0 | 2.6 | 3.2 | 2.2 | |
| IM-ALL | (%) | 2.5 | 1.7 | 1.2 | 1.0 | 1.5 | 1.4 | |
| BI-ALL | (%) | −0.9 | −0.6 | 1.3 | 0.5 | 1.6 | 0.7 | |
| CN | (nmol/L) | 0.29 | 0.57 | 1.15 | 5.74 | 34.2 | 80.8 | |
| IM-IN | (%) | 19.4 | 13.4 | 9.0 | 7.1 | 6.6 | 6.8 | |
| IM-ALL | (%) | 6.3 | 7.7 | 5.1 | 3.6 | 4.8 | 4.8 | |
| BI-ALL | (%) | 34.5 | 13.4 | 4.8 | −0.3 | 1.0 | −1.4 | |
| ALDO | (nmol/L) | 0.013 | 0.026 | 0.051 | 0.255 | 0.699 | 2.430 | |
| IM-IN | (%) | n.a. | n.a. | 25.4 | 4.2 | 7.2 | 5.5 | |
| IM-ALL | (%) | n.a. | n.a. | 9.1 | 3.7 | 3.2 | 2.9 | |
| BI-ALL | (%) | n.a. | n.a. | 17.3 | 6.2 | 2.9 | 14.1 |
| Analyte | Unit | F1 | F2 | F3 | F4 | F5 | M1 | M2 | M3 | M4 | M5 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 11-DOC | (nmol/L) | 0.25 | 0.20 | 0.16 | 0.10 | 0.10 | 0.24 | 0.16 | 0.14 | 0.09 | 0.06 |
| (%) | 5.4 | 10.8 | 7.4 | 13.5 | 18.9 | 13.4 | 5.7 | 9.0 | 12.0 | 9.2 | |
| 17P | (nmol/L) | 2.24 | 2.15 | 2.42 | 1.10 | 0.87 | 1.29 | 1.46 | 2.61 | 2.40 | 2.34 |
| (%) | 1.8 | 6.2 | 4.6 | 4.5 | 13.0 | 8.7 | 2.5 | 3.6 | 1.3 | 1.2 | |
| ANDRO | (nmol/L) | 0.35 | 4.54 | 4.03 | 2.18 | 1.83 | 0.25 | 1.66 | 3.08 | 2.56 | 1.84 |
| (%) | 2.6 | 5.9 | 4.5 | 3.0 | 12.9 | 11.2 | 1.8 | 2.6 | 0.8 | 1.0 | |
| DHEA | (nmol/L) | 3.68 | 17.59 | 17.89 | 9.87 | 6.21 | 2.68 | 9.52 | 14.58 | 8.09 | 4.51 |
| (%) | 45.0 | 7.3 | 7.4 | 5.5 | 12.6 | 22.1 | 6.3 | 7.4 | 11.5 | 23.9 | |
| DHT | (nmol/L) | n.a. | 0.231 | 0.283 | 0.138 | 0.120 | 0.140 | 0.569 | 1.245 | 0.960 | 0.940 |
| (%) | n.a. | 23.9 | 12.3 | 16.9 | 16.4 | 24.7 | 7.4 | 7.7 | 6.6 | 10.9 | |
| PROG | (nmol/L) | 46.12 | 3.98 | 23.57 | 1.70 | 0.13 | 28.89 | 0.14 | 0.25 | 0.20 | 0.19 |
| (%) | 0.6 | 5.8 | 4.9 | 3.9 | 7.5 | 9.0 | 5.9 | 4.4 | 5.8 | 4.3 | |
| TES | (nmol/L) | 0.07 | 0.97 | 1.06 | 0.68 | 0.76 | 0.49 | 7.60 | 18.49 | 16.01 | 14.18 |
| (%) | 10.5 | 7.9 | 5.9 | 5.8 | 13.5 | 11.1 | 1.6 | 3.1 | 2.1 | 2.2 | |
| 21-DF | (nmol/L) | 0.03 | 0.06 | 0.06 | 0.07 | 0.08 | 0.02 | 0.05 | 0.04 | 0.07 | 0.08 |
| (%) | 38.5 | 19.9 | 40.4 | 18.9 | 22.6 | 35.1 | 47.6 | 28.0 | 35.4 | 32.3 | |
| 11-DF | (nmol/L) | 1.23 | 1.52 | 0.72 | 0.81 | 0.96 | 0.78 | 1.48 | 0.87 | 1.02 | 0.85 |
| (%) | 3.4 | 6.9 | 5.4 | 2.5 | 11.6 | 10.4 | 4.9 | 4.4 | 2.4 | 3.4 | |
| CC | (nmol/L) | 5.13 | 15.18 | 11.93 | 16.27 | 10.65 | 6.65 | 7.91 | 15.78 | 10.29 | 7.36 |
| (%) | 4.6 | 6.8 | 5.7 | 5.2 | 12.1 | 7.1 | 5.2 | 9.6 | 4.6 | 4.3 | |
| DHEAS | (nmol/L) | 451 | 4283 | 4307 | 2013 | 1407 | 419 | 4153 | 6115 | 3189 | 1744 |
| (%) | 1.3 | 5.1 | 4.4 | 3.2 | 11.7 | 7.6 | 1.5 | 1.9 | 1.0 | 0.7 | |
| E2 | (nmol/L) | 0.393 | 0.391 | 2.365 | 0.102 | 0.027 | 0.104 | 0.029 | 0.093 | 0.080 | 0.079 |
| (%) | 4.3 | 5.1 | 5.9 | 7.9 | 10.9 | 11.0 | 12.4 | 5.3 | 8.0 | 5.9 | |
| CC | (nmol/L) | 187 | 355 | 358 | 401 | 406 | 223 | 247 | 343 | 361 | 313 |
| (%) | 1.8 | 5.2 | 5.3 | 3.5 | 11.1 | 10.2 | 4.0 | 2.6 | 1.9 | 3.2 | |
| CL | (nmol/L) | 47.3 | 68.7 | 51.8 | 48.3 | 49.1 | 54.2 | 54.3 | 52.0 | 51.6 | 41.1 |
| (%) | 3.9 | 7.5 | 6.6 | 4.6 | 12.1 | 10.3 | 6.5 | 7.4 | 6.1 | 5.7 | |
| ALDO | (nmol/L) | 0.271 | 0.302 | 0.239 | 0.214 | 0.228 | 0.274 | 0.243 | 0.176 | 0.146 | 0.189 |
| (%) | 7.1 | 6.6 | 8.2 | 8.1 | 11.0 | 8.8 | 5.6 | 10.3 | 5.2 | 5.7 |
| Comparator Assay † | Analyte | Intercept | (95% CI) of Intercept | Slope | (95% CI) of Slope | R2 * | (95% CI) of R2 | Linear § | n |
|---|---|---|---|---|---|---|---|---|---|
| ECLIA | CL | 0.84 | (−13.51 to 10.00) | 1.03 | (1.00 to 1.08) | 0.966 | (0.948 to 0.978) | Yes | 86 |
| ECLIA | DHEAS | −103.84 | (−206.60 to 11.90) | 0.84 | (0.81 to 0.86) | 0.974 | (0.963 to 0.983) | Yes | 107 |
| ECLIA | E2 | 0.00 | (−0.02 to 0.02) | 1.04 | (0.99 to 1.16) | 0.891 | (0.835 to 0.928) | Yes | 82 |
| ECLIA | PROG | −0.57 | (−0.68 to −0.46) | 1.03 | (0.99 to 1.08) | 0.937 | (0.892 to 0.964) | Yes | 51 |
| ECLIA | TES | 0.05 | (−0.01 to 0.13) | 1.03 | (1.00 to 1.06) | 0.959 | (0.939 to 0.972) | Yes | 96 |
| ECLIA | 17P | 0.02 | (−0.28 to 0.33) | 0.39 | (0.31 to 0.52) | 0.670 | (0.528 to 0.776) | Yes | 79 |
| CLIA | ANDRO | −0.03 | (−0.29 to 0.22) | 0.52 | (0.49 to 0.55) | 0.936 | (0.903 to 0.958) | Yes | 84 |
| ELISA | DHEA | 1.09 | (−0.34 to 2.30) | 0.72 | (0.62 to 0.82) | 0.897 | (0.844 to 0.933) | Yes | 79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, V.; Stuppner, H.; Risch, L.; Seger, C. Design and Validation of a Sensitive Multisteroid LC-MS/MS Assay for the Routine Clinical Use: One-Step Sample Preparation with Phospholipid Removal and Comparison to Immunoassays. Int. J. Mol. Sci. 2022, 23, 14691. https://doi.org/10.3390/ijms232314691
Braun V, Stuppner H, Risch L, Seger C. Design and Validation of a Sensitive Multisteroid LC-MS/MS Assay for the Routine Clinical Use: One-Step Sample Preparation with Phospholipid Removal and Comparison to Immunoassays. International Journal of Molecular Sciences. 2022; 23(23):14691. https://doi.org/10.3390/ijms232314691
Chicago/Turabian StyleBraun, Valentin, Hermann Stuppner, Lorenz Risch, and Christoph Seger. 2022. "Design and Validation of a Sensitive Multisteroid LC-MS/MS Assay for the Routine Clinical Use: One-Step Sample Preparation with Phospholipid Removal and Comparison to Immunoassays" International Journal of Molecular Sciences 23, no. 23: 14691. https://doi.org/10.3390/ijms232314691