
The Innate Immune System in Cardiovascular Diseases and Its Role in Doxorubicin-Induced Cardiotoxicity

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
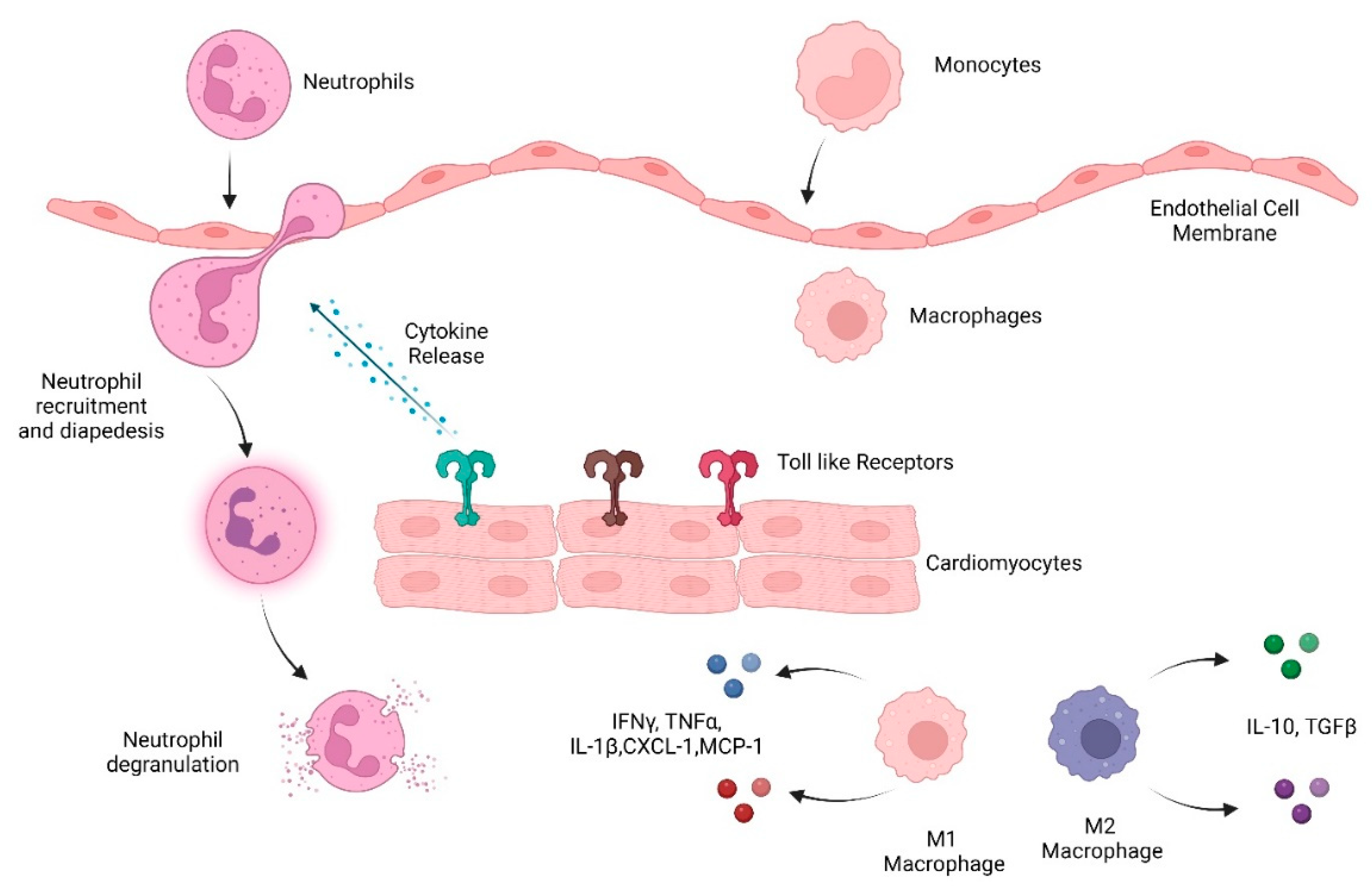
2. Inflammation in the Heart
2.1. Pattern-Recognition Receptors
- Toll-like Receptors
- TLRs in the heart
- Inflammasome
2.2. Cytokines
- Pro-inflammatory cytokines
- Anti-inflammatory cytokines
- Chemokines
2.3. Neutrophils in Heart Damage
- Neutrophils in CVDs
- Neutrophil Elastase
- Neutrophil elastase in CVDs
2.4. Macrophages in CVD
- Cardiac macrophage heterogeneity and diversity
- Role of macrophages in cardiac damage and repair
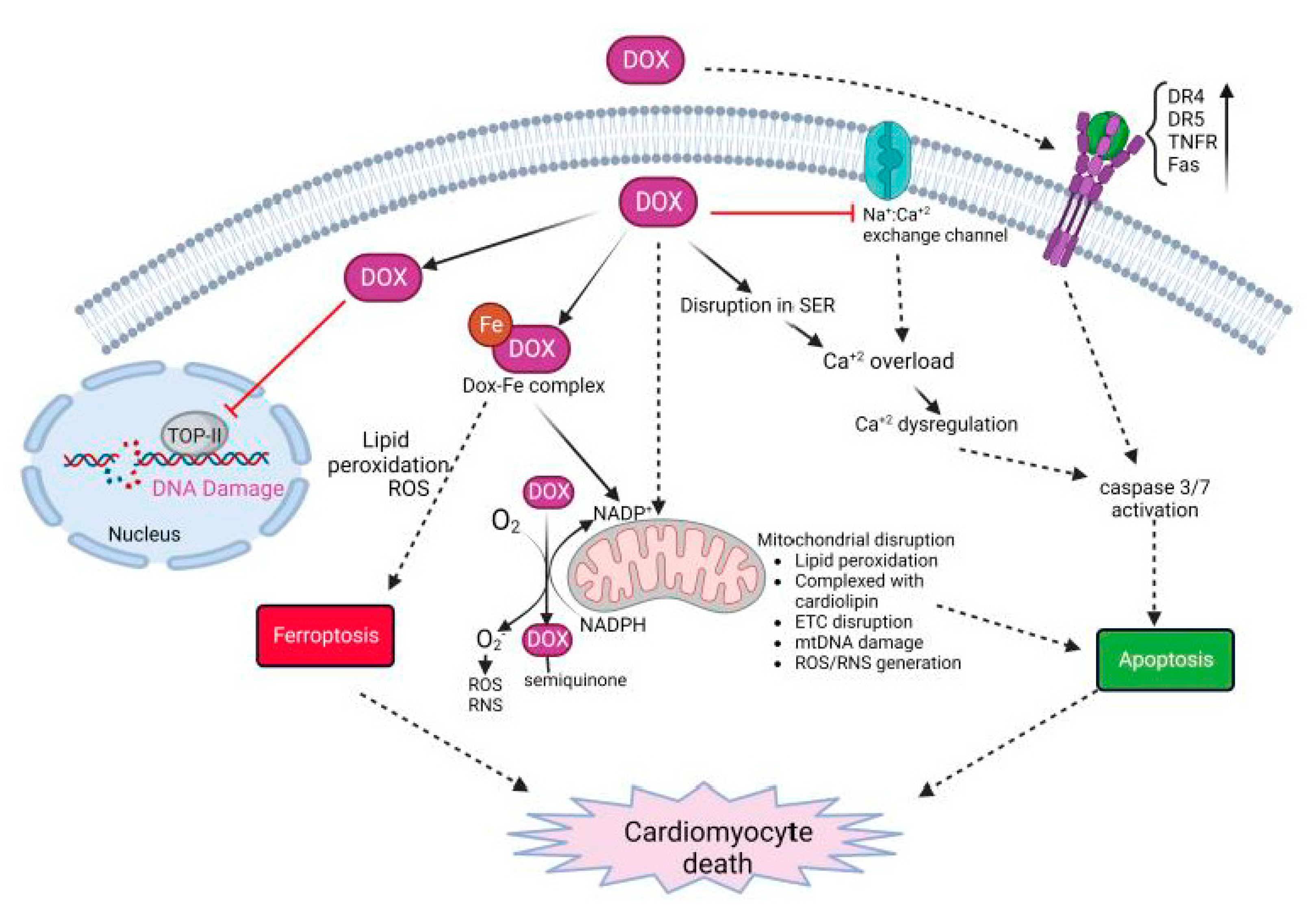
3. Dox-Induced Cardiomyocyte Injury
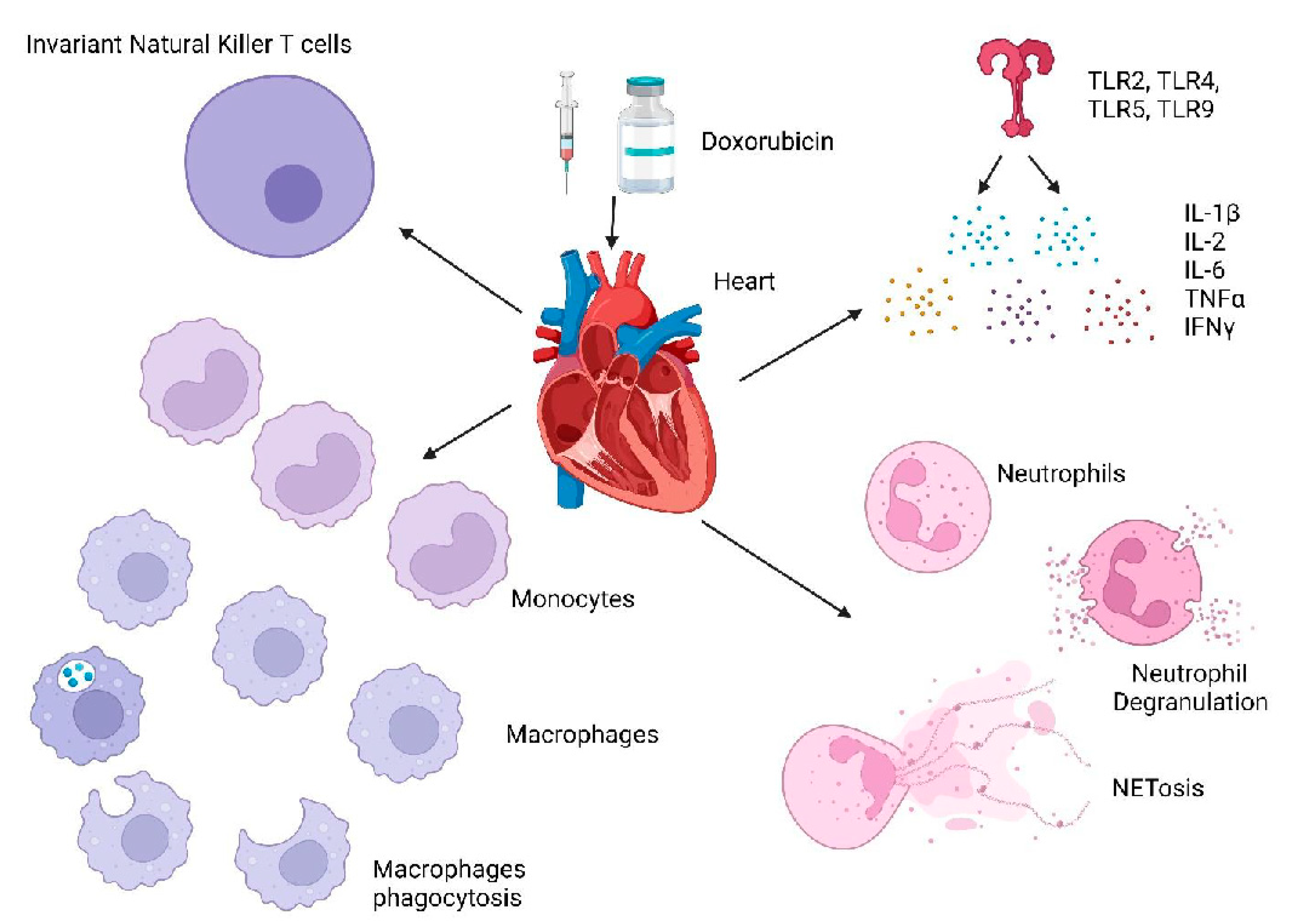
3.1. Role of Immune Response in Dox-Induced Cardiotoxicity
3.1.1. Cytokines/Chemokines
3.1.2. TLRs
3.1.3. Innate Immune Cells
- Neutrophils
- Macrophages
- Invariant natural killer T cells
- Conclusion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| Dox | Doxorubicin |
| CVDs | Cardiovascular diseases |
| MI | Myocardial infarction |
| DAMPs | Damage-associated molecular patterns |
| PRRs | Pattern-recognition receptors |
| TLRs | Toll-like receptors |
| NOD | Nucleotide-binding and oligomerization domain |
| IL-1 | Interleukin-1 |
| TNFα | Tumor necrosis factor α |
| G-CSF | Granulocyte colony-stimulating factor |
| ECM | Extracellular matrix |
| HMGB1 | High-mobility group box protein 1 |
| IRAK | IL-1 receptor-associated kinase |
| TRAF | TNF-receptor-associated factor |
| TAK | Transforming growth factor β-activated kinase |
| IKK Complex | IκB complex |
| TBK1 | TANK Binding Kinase-1 |
| IRF3 | Interferon-regulatory factor-3 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| NLRP3 | NOD-, LRR- and pyrin-domain-containing protein 3 |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| MMPs | Matrix metalloproteinases |
| ICAM-1 | Intercellular adhesion molecule |
| VCAM-1 | Vascular cell adhesion molecule |
| TIMP | Tissue inhibitor of metalloproteinases |
| TGFβ | Tumor growth factor-β |
| CXCL1 | C-X-C Motif Chemokine Ligand 1 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| CCL2 | C-C motif ligand 2 |
| CCR2 | C-C motif receptor 2 |
| CXCR4 | C-X-C Motif chemokine receptor 4 |
| ROS | Reactive oxygen species |
| NE | Neutrophil elastase |
| PI3K/AKT | Phosphatidylinositol 3-kinase/protein kinase B |
| ERK | Extracellular signal-regulated kinases |
| JNK | c-Jun N-terminal kinases |
| MAPK | Mitogen-activated protein kinases |
| MHC | Major histocompatibility complex |
| HSPs | Heat-shock proteins |
| ATP | Adenosine triphosphate |
| PAMPs | Pathogen-associated molecular patterns |
| PDGFs | Platelet-derived growth factors |
| IGF-1 | Insulin-like growth factors-1 |
| VEGF | Vascular endothelial growth factor-1 |
| DR4 and 5 | Death receptors 4 and 5 |
| AMPK | AMP-activated protein kinase |
| MIF | Macrophage migration inhibitory factor |
| NT-proBNP | NT-proB-type Natriuretic peptide |
| NOX2 | NADPH oxidase 2 |
| WT | Wild type |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| CX3CR1 | C-X3-C Motif Chemokine Receptor 1 |
| iNKT | Invariant natural killer T-cells |
| αGC | Alpha-galactosylceramide |
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- WHO. Cardiovascular Disease (CVDs). 2021. Available online: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 23 October 2022).
- Goldsborough, E.; Osuji, N.; Blaha, M.J. Assessment of Cardiovascular Disease Risk: A 2022 Update. Endocrinol. Metab. Clin. 2022, 51, 483–509. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Noels, H.; Weber, C.; Koenen, R.R. Chemokines as Therapeutic Targets in Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2019, 39, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Tocchetti, C.G.; Ameri, P.; De Boer, R.A.; D’Alessandra, Y.; Russo, M.; Sorriento, D.; Ciccarelli, M.; Kiss, B.; Bertrand, L.; Dawson, D.; et al. Cardiac dysfunction in cancer patients: Beyond direct cardiomyocyte damage of anticancer drugs: Novel cardio-oncology insights from the joint 2019 meeting of the ESC Working Groups of Myocardial Function and Cellular Biology of the Heart. Cardiovasc. Res. 2020, 116, 1820–1834. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Bhatia, S. Genetics of Anthracycline Cardiomyopathy in Cancer Survivors. JACC CardioOncology 2020, 2, 539–552. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, A.; Urbanek, K.; Cappetta, D.; Piegari, E.; Ciuffreda, L.P.; Rivellino, A.; Russo, R.; Esposito, G.; Rossi, F.; Berrino, L. Doxorubicin cardiotoxicity and target cells: A broader perspective. Cardio-Oncology 2016, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- van Nieuwenhoven, F.A.; Turner, N.A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharmacol. 2013, 58, 182–188. [Google Scholar] [CrossRef]
- Baci, D.; Bosi, A.; Parisi, L.; Buono, G.; Mortara, L.; Ambrosio, G.; Bruno, A. Innate Immunity Effector Cells as Inflammatory Drivers of Cardiac Fibrosis. Int. J. Mol. Sci. 2020, 21, 7165. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Liu, L.; Kelley, J.; Kao, R.; Williams, D.; Li, C. Toll-Like Receptors: New Players in Myocardial Ischemia/Reperfusion Injury. Antioxidants Redox Signal. 2011, 15, 1875–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, W. Toll-like receptor signaling: A critical modulator of cell survival and ischemic injury in the heart. Am. J. Physiol. Circ. Physiol. 2009, 296, H1–H12. [Google Scholar] [CrossRef] [Green Version]
- Vilahur, G.; Badimon, L. Ischemia/reperfusion activates myocardial innate immune response: The key role of the toll-like receptor. Front. Physiol. 2014, 5, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyama, J.-I.; Blais, C.; Liu, X.; Pu, M.; Kobzik, L.; Kelly, R.A.; Bourcier, T. Reduced Myocardial Ischemia-Reperfusion Injury in Toll-Like Receptor 4-Deficient Mice. Circulation 2004, 109, 784–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzaroma, E.; Abbate, A.; Toldo, S. NLRP3 Inflammasome Inhibitors in Cardiovascular Diseases. Molecules 2021, 26, 976. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 Inflammasome in Acute Myocardial Infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef]
- Sivasubramanian, N.; Coker, M.L.; Kurrelmeyer, K.M.; MacLellan, W.R.; DeMayo, F.; Spinale, F.G.; Mann, D.L. Left Ventricular Remodeling in Transgenic Mice With Cardiac Restricted Overexpression of Tumor Necrosis Factor. Circulation 2001, 104, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Mattila, P.; Majuri, M.-L.; Renkonen, R. TNFa-Induced Expression of Endothelial Adhesion Molecules, ICAM-1 and VCAM-1, is Linked to Protein Kinase C Activation. Scand. J. Immunol. 1992, 36, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Frangogiannis, N.G. The role of IL-1 in the pathogenesis of heart disease. Arch. Immunol. Ther. Exp. 2009, 57, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Ma, C.; Yang, H.; Zhang, P.-Y. Transforming growth factor β and its role in heart disease. Exp. Ther. Med. 2017, 13, 2123–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-L.; Yin, R.; Wang, S.-N.; Ying, R. A Review of CXCL1 in Cardiac Fibrosis. Front. Cardiovasc. Med. 2021, 8, 674498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Cao, H.-J.; Han, X.; Teng, F.; Chen, C.; Yang, J.; Yan, X.; Li, P.-B.; Liu, Y.; Xia, Y.-L.; et al. Chemokine Receptor CXCR-2 Initiates Atrial Fibrillation by Triggering Monocyte Mobilization in Mice. Hypertension 2020, 76, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Sreejit, G.; Johnson, J.; Jaggers, R.M.; Dahdah, A.; Murphy, A.J.; Hanssen, N.M.J.; Nagareddy, P.R. Neutrophils in cardiovascular disease: Warmongers, peacemakers, or both? Cardiovasc. Res. 2021, 118, 2596–2609. [Google Scholar] [CrossRef]
- Stroka, K.M.; Levitan, I.; Aranda-Espinoza, H. OxLDL and substrate stiffness promote neutrophil transmigration by enhanced endothelial cell contractility and ICAM-1. J. Biomech. 2012, 45, 1828–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soehnlein, O.; Zernecke, A.; Eriksson, E.E.; Rothfuchs, A.G.; Pham, C.T.; Herwald, H.; Bidzhekov, K.; Rottenberg, M.E.; Weber, C.; Lindbom, L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 2008, 112, 1461–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Sreejit, G.; Abo-Aly, M.; Jaggers, R.M.; Chelvarajan, L.; Johnson, J.; Pernes, G.; Athmanathan, B.; Abdel-Latif, A.; Murphy, A.J. NETosis Is Required for S100A8/A9-Induced Granulopoiesis After Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2020, 40, 2805–2807. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, S.; Weng, X.; Zeng, S.; Yu, L.; Guo, J.; Xu, Y. Brg1 deficiency in vascular endothelial cells blocks neutrophil recruitment and ameliorates cardiac ischemia-reperfusion injury in mice. Int. J. Cardiol. 2018, 269, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Vajen, T.; Koenen, R.R.; Werner, I.; Staudt, M.; Projahn, D.; Curaj, A.; Sönmez, T.T.; Simsekyilmaz, S.; Schumacher, D.; Möllmann, J.; et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci. Rep. 2018, 8, 10647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.-P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127, e232–e249. [Google Scholar] [CrossRef]
- Gramegna, A.; Amati, F.; Terranova, L.; Sotgiu, G.; Tarsia, P.; Miglietta, D.; Calderazzo, M.A.; Aliberti, S.; Blasi, F. Neutrophil elastase in bronchiectasis. Respir. Res. 2017, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Chua, F. Neutrophil Elastase: Mediator of Extracellular Matrix Destruction and Accumulation. Proc. Am. Thorac. Soc. 2006, 3, 424–427. [Google Scholar] [CrossRef]
- Zhang, N.; Aiyasiding, X.; Li, W.-J.; Liao, H.-H.; Tang, Q.-Z. Neutrophil degranulation and myocardial infarction. Cell Commun. Signal. 2022, 20, 50. [Google Scholar] [CrossRef]
- Ogura, Y.; Tajiri, K.; Murakoshi, N.; Xu, D.; Yonebayashi, S.; Li, S.; Okabe, Y.; Feng, D.; Shimoda, Y.; Song, Z.; et al. Neutrophil Elastase Deficiency Ameliorates Myocardial Injury Post Myocardial Infarction in Mice. Int. J. Mol. Sci. 2021, 22, 722. [Google Scholar] [CrossRef]
- Houghton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Pan, J.; Sun, Y.; Zong, S.; Zhang, R.; Li, Y.; Yu, Z.; Liu, J.; Zang, S. Increased Neutrophil elastase and proteinase 3 are closely associated with occurrence and severity of stroke and acute myocardial infarction in patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2022, 186, 109853. [Google Scholar] [CrossRef]
- Coeshott, C.; Ohnemus, C.; Pilyavskaya, A.; Ross, S.; Wieczorek, M.; Kroona, H.; Leimer, A.H.; Cheronis, J. Converting enzyme-independent release of tumor necrosis factor α and IL-1β from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc. Natl. Acad. Sci. USA 1999, 96, 6261–6266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendergraft, W.F.; Rudolph, E.H.; Falk, R.J.; Jahn, J.E.; Grimmler, M.; Hengst, L.; Jennette, J.C.; Preston, G.A. Proteinase 3 sidesteps caspases and cleaves p21Waf1/Cip1/Sdi1 to induce endothelial cell apoptosis. Kidney Int. 2004, 65, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, G.A.; Zarella, C.S.; Pendergraft, W.F.; Rudolph, E.H.; Yang, J.J.; Sekura, S.B.; Jennette, J.C.; Falk, R.J. Novel Effects of Neutrophil-Derived Proteinase 3 and Elastase on the Vascular Endothelium Involve In Vivo Cleavage of NF- B and Proapoptotic Changes in JNK, ERK, and p38 MAPK Signaling Pathways. J. Am. Soc. Nephrol. 2002, 13, 2840–2849. [Google Scholar] [CrossRef] [Green Version]
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.J. Macrophages in Cardiovascular Homeostasis and Disease. Circulation 2018, 138, 2452–2455. [Google Scholar] [CrossRef]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Williams, J.W.; Giannarelli, C.; Rahman, A.; Randolph, G.J.; Kovacic, J.C. Macrophage Biology, Classification, and Phenotype in Cardiovascular Disease: JACC Macrophage in CVD Series (Part 1). J. Am. Coll. Cardiol. 2018, 72, 2166–2180. [Google Scholar] [CrossRef]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, N.I.; Goldschmidt-Clermont, P.J.; Parker-Thornburg, J.; Shapiro, S.D.; Kolattukudy, P.E. Contribution of monocytes/macrophages to compensatory neovascularization: The drilling of metalloelastase-positive tunnels in ischemic myocardium. Circ. Res. 2000, 87, 378–384. [Google Scholar] [CrossRef] [Green Version]
- Rehman, J.; Li, J.; Orschell, C.M.; March, K.L. Peripheral Blood “Endothelial Progenitor Cells” Are Derived From Monocyte/Macrophages and Secrete Angiogenic Growth Factors. Circulation 2003, 107, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- van Amerongen, M.J.; Harmsen, M.C.; van Rooijen, N.; Petersen, A.H.; van Luyn, M.J. Macrophage Depletion Impairs Wound Healing and Increases Left Ventricular Remodeling after Myocardial Injury in Mice. Am. J. Pathol. 2007, 170, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 2715. [Google Scholar] [CrossRef]
- Gao, Y.; Qian, N.; Xu, J.; Wang, Y. The Roles of Macrophages in Heart Regeneration and Repair After Injury. Front. Cardiovasc. Med. 2021, 8, 744615. [Google Scholar] [CrossRef]
- Deberge, M.; Yeap, X.Y.; Dehn, S.; Zhang, S.; Grigoryeva, L.; Misener, S.; Procissi, D.; Zhou, X.; Lee, D.; Muller, W.A.; et al. MerTK Cleavage on Resident Cardiac Macrophages Compromises Repair After Myocardial Ischemia Reperfusion Injury. Circ. Res. 2017, 121, 930–940. [Google Scholar] [CrossRef]
- Howangyin, K.-Y. Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation 2016, 133, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Song, L.; Wang, L.; Yukht, A.; Ruther, H.; Li, F.; Qin, M.; Ghiasi, H.; Sharifi, B.G.; Shah, P.K. Deficiency of GATA3-Positive Macrophages Improves Cardiac Function Following Myocardial Infarction or Pressure Overload Hypertrophy. J. Am. Coll. Cardiol. 2018, 72, 885–904. [Google Scholar] [CrossRef] [PubMed]
- Leor, J.; Rozen, L.; Zuloff-Shani, A.; Feinberg, M.S.; Amsalem, Y.; Barbash, I.M.; Kachel, E.; Holbova, R.; Mardor, Y.; Daniels, D.; et al. Ex Vivo Activated Human Macrophages Improve Healing, Remodeling, and Function of the Infarcted Heart. Circulation 2006, 114, I-94–I-100. [Google Scholar] [CrossRef]
- Olson, R.D.; Gambliel, H.A.; Vestal, R.E.; Shadle, S.E.; Charlier, H.A.; Cusack, B.J. Doxorubicin Cardiac Dysfunction: Effects on Calcium Regulatory Proteins, Sarcoplasmic Reticulum, and Triiodothyronine. Cardiovasc. Toxicol. 2005, 5, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. IJC Heart Vasc. 2015, 10, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Ni, C.; Ma, P.; Wang, R.; Lou, X.; Liu, X.; Qin, Y.; Xue, R.; Blasig, I.; Erben, U.; Qin, Z. Doxorubicin-induced cardiotoxicity involves IFNγ-mediated metabolic reprogramming in cardiomyocytes. J. Pathol. 2019, 247, 320–332. [Google Scholar] [CrossRef]
- Ma, P.; Qin, Y.; Cao, H.; Erben, U.; Ni, C.; Qin, Z. Temporary blockade of interferon-γ ameliorates doxorubicin-induced cardiotoxicity without influencing the anti-tumor effect. Biomed. Pharmacother. 2020, 130, 110587. [Google Scholar] [CrossRef]
- Yu, L.-R.; Cao, Z.; Makhoul, I.; Daniels, J.R.; Klimberg, S.; Wei, J.Y.; Bai, J.P.; Li, J.; Lathrop, J.T.; Beger, R.D.; et al. Immune response proteins as predictive biomarkers of doxorubicin-induced cardiotoxicity in breast cancer patients. Exp. Biol. Med. 2017, 243, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Rassaf, T.; Weber, C.; Bernhagen, J. Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2014, 102, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Pang, J.; Chen, Y.; Bucala, R.; Zhang, Y.; Ren, J. Macrophage Migration Inhibitory Factor (MIF) Deficiency Exacerbates Aging-Induced Cardiac Remodeling and Dysfunction Despite Improved Inflammation: Role of Autophagy Regulation. Sci. Rep. 2016, 6, 22488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-γ-Inducible Protein 10 (IP-10; CXCL10)-Deficient Mice Reveal a Role for IP-10 in Effector T Cell Generation and Trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, Z.S.; Brunt, V.E.; Hutton, D.A.; Casso, A.G.; Ziemba, B.P.; Melov, S.; Campisi, J.; Seals, D.R. Tumor Necrosis Factor Alpha-Mediated Inflammation and Remodeling of the Extracellular Matrix Underlies Aortic Stiffening Induced by the Common Chemotherapeutic Agent Doxorubicin. Hypertension 2021, 77, 1581–1590. [Google Scholar] [CrossRef]
- Alves, M.T.; Simões, R.; Pestana, R.M.C.; de Oliveira, A.N.; Oliveira, H.H.M.; Soares, C.E.; Sabino, A.D.P.; Silva, L.M.; Gomes, K.B. Interleukin-10 Levels are Associated with Doxorubicin-Related Cardiotoxicity in Breast Cancer Patients in a One-Year Follow-Up Study. Immunol. Investig. 2021, 51, 883–898. [Google Scholar] [CrossRef]
- Ma, Z.-G.; Kong, C.-Y.; Wu, H.-M.; Song, P.; Zhang, X.; Yuan, Y.-P.; Deng, W.; Tang, Q.-Z. Toll-like receptor 5 deficiency diminishes doxorubicin-induced acute cardiotoxicity in mice. Theranostics 2020, 10, 11013–11025. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.Z.; Hu, J.X.; Wu, H.; Li, Y.L.; Chen, H.L.; Bai, H.; Hai, C.X. ROS-activated p38 MAPK/ERK-Akt cascade plays a central role in palmitic acid-stimulated hepatocyte proliferation. Free Radic. Biol. Med. 2011, 51, 539–551. [Google Scholar] [CrossRef]
- Guo, Z.; Tang, N.; Liu, F.; Yang, Z.; Ma, S.; An, P.; Wu, H.; Fan, D.; Tang, Q. TLR9 deficiency alleviates doxorubicin-induced cardiotoxicity via the regulation of autophagy. J. Cell. Mol. Med. 2020, 24, 10913–10923. [Google Scholar] [CrossRef]
- Nozaki, N.; Shishido, T.; Takeishi, Y.; Kubota, I. Modulation of Doxorubicin-Induced Cardiac Dysfunction in Toll-Like Receptor-2–Knockout Mice. Circulation 2004, 110, 2869–2874. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Xinyong, C.; Hongmin, Z.; Jing, W.; Lang, H.; Ping, Z. TLR2 and TLR3 expression as a biomarker for the risk of doxorubicin-induced heart failure. Toxicol. Lett. 2018, 295, 205–211. [Google Scholar] [CrossRef]
- Ye, S.; Su, L.; Shan, P.; Ye, B.; Wu, S.; Liang, G.; Huang, W. LCZ696 Attenuated Doxorubicin-Induced Chronic Cardiomyopathy Through the TLR2-MyD88 Complex Formation. Front. Cell Dev. Biol. 2021, 9, 654051. [Google Scholar] [CrossRef]
- Riad, A.; Bien, S.; Gratz, M.; Escher, F.; Heimesaat, M.M.; Bereswill, S.; Krieg, T.; Felix, S.B.; Schultheiss, H.P.; Kroemer, H.K.; et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur. J. Heart Fail. 2008, 10, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Q.; Qi, H.; Wang, C.; Wang, C.; Zhang, J.; Dong, L. Doxorubicin-Induced Systemic Inflammation Is Driven by Upregulation of Toll-Like Receptor TLR4 and Endotoxin Leakage. Cancer Res. 2016, 76, 6631–6642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, S.; Wang, Y.; Ogawa, H.; Horitani, K.; Sano, M.; Polizio, A.H.; Kour, A.; Yura, Y.; Doviak, H.; Walsh, K. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.-H.; Contreras, G.P.; Yeh, T.-Y. Potential Role of Neutrophil Extracellular Traps in Cardio-Oncology. Int. J. Mol. Sci. 2022, 23, 3573. [Google Scholar] [CrossRef]
- Bhagat, A.; Shrestha, P.; Jeyabal, P.; Peng, Z.; Watowich, S.S.; Kleinerman, E.S. Doxorubicin-induced cardiotoxicity is mediated by neutrophils through release of neutrophil elastase. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Krysko, O.; Heyndrickx, L.; Aaes, T.L.; Delvaeye, T.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. TNF/TNF-R1 pathway is involved in doxorubicin-induced acute sterile inflammation. Cell Death Dis. 2013, 4, e961. [Google Scholar] [CrossRef] [Green Version]
- Krysko, D.; Kaczmarek, A.; Krysko, O.; Heyndrickx, L.; Woznicki, J.; Bogaert, P.; Cauwels, A.; Takahashi, N.; Magez, S.; Bachert, C.; et al. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ. 2011, 18, 1316–1325. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lv, J.; Jiang, S.; Ma, Z.; Wang, D.; Hu, W.; Deng, C.; Fan, C.; Di, S.; Sun, Y.; et al. The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death Dis. 2016, 7, e2234. [Google Scholar] [CrossRef] [Green Version]
- Katare, P.B.; Nizami, H.L.; Paramesha, B.; Dinda, A.K.; Banerjee, S.K. Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci. Rep. 2020, 10, 19232. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, X.; Bao, H.; Mi, S.; Cai, W.; Yan, H.; Wang, Q.; Wang, Z.; Yan, J.; Fan, G.; et al. Toll-Like Receptor (TLR) 2 and TLR4 Differentially Regulate Doxorubicin Induced Cardiomyopathy in Mice. PLoS ONE 2012, 7, e40763. [Google Scholar] [CrossRef]
- Boyd, J.H.; Mathur, S.; Wang, Y.; Bateman, R.M.; Walley, K.R. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-κB dependent inflammatory response☆. Cardiovasc. Res. 2006, 72, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Cao, Z.; Wang, M.; Liu, X.; Gao, T.; Hu, Q.; Yuan, W.; Lin, L. Up-regulated TLR 4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction. J. Cell. Mol. Med. 2015, 19, 2728–2740. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; AlThagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2018, 20, 29–39. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, A.; Sun, X.; Yang, Y.; Zhang, L.; Bai, H.; Ben, J.; Zhu, X.; Li, X.; Wang, Z.; et al. Self-Maintenance of Cardiac Resident Reparative Macrophages Attenuates Doxorubicin-Induced Cardiomyopathy Through the SR-A1-c-Myc Axis. Circ. Res. 2020, 127, 610–627. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Wang, Y.; Xu, Y.; Wang, Z.; Liu, L.; Wang, M.; Ye, D.; Zhang, J.; Yang, Z.; Lin, Y.; et al. Interleukin-22 deficiency alleviates doxorubicin-induced oxidative stress and cardiac injury via the p38 MAPK/macrophage/Fizz3 axis in mice. Redox Biol. 2020, 36, 101636. [Google Scholar] [CrossRef]
- Ye, J.; Huang, Y.; Que, B.; Chang, C.; Liu, W.; Hu, H.; Liu, L.; Shi, Y.; Wang, Y.; Wang, M.; et al. Interleukin-12p35 Knock Out Aggravates Doxorubicin-Induced Cardiac Injury and Dysfunction by Aggravating the Inflammatory Response, Oxidative Stress, Apoptosis and Autophagy in Mice. eBioMedicine 2018, 35, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Liu, Y.; Tang, H.; Qiu, M.; Li, C.; Duan, C.; Wang, C.; Yang, J.; Zhou, X. Glabridin Prevents Doxorubicin-Induced Cardiotoxicity Through Gut Microbiota Modulation and Colonic Macrophage Polarization in Mice. Front. Pharmacol. 2019, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Usui, F.; Karasawa, T.; Kawashima, A.; Kimura, H.; Mizushina, Y.; Shirasuna, K.; Mizukami, H.; Kasahara, T.; Hasebe, N.; et al. NLRP3 Deficiency Reduces Macrophage Interleukin-10 Production and Enhances the Susceptibility to Doxorubicin-induced Cardiotoxicity. Sci. Rep. 2016, 6, 26489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, D.; Johnson, T.; Dargani, Z.T. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells 2019, 8, 1224. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, M.; Zhong, C.; Xu, B.; Kang, L. M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer. Biomater. Res. 2022, 26, 14. [Google Scholar] [CrossRef] [PubMed]
- Obata, Y.; Ishimori, N.; Saito, A.; Kinugawa, S.; Yokota, T.; Takada, S.; Nakano, I.; Kakutani, N.; Yamanashi, K.; Anzai, T. Activation of invariant natural killer T cells by alpha-galactosylceramide ameliorates doxorubicin-induced cardiotoxicity in mice. Eur. J. Prev. Cardiol. 2020, 27, 2358–2361. [Google Scholar] [CrossRef]
- Safra, T.; Muggia, F.; Jeffers, S.; Tsao-Wei, D.D.; Groshen, S.; Lyass, O.; Henderson, R.; Berry, G.; Gabizon, A. Pegylated liposomal doxorubicin (doxil): Reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann. Oncol. 2000, 11, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Gyöngyösi, M.; Lukovic, D.; Zlabinger, K.; Spannbauer, A.; Gugerell, A.; Pavo, N.; Traxler, D.; Pils, D.; Maurer, G.; Jakab, A.; et al. Liposomal doxorubicin attenuates cardiotoxicity via induction of interferon-related DNA damage resistance. Cardiovasc. Res. 2019, 116, 970–982. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.A.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhagat, A.; Shrestha, P.; Kleinerman, E.S. The Innate Immune System in Cardiovascular Diseases and Its Role in Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 14649. https://doi.org/10.3390/ijms232314649
Bhagat A, Shrestha P, Kleinerman ES. The Innate Immune System in Cardiovascular Diseases and Its Role in Doxorubicin-Induced Cardiotoxicity. International Journal of Molecular Sciences. 2022; 23(23):14649. https://doi.org/10.3390/ijms232314649
Chicago/Turabian StyleBhagat, Anchit, Pradeep Shrestha, and Eugenie S. Kleinerman. 2022. "The Innate Immune System in Cardiovascular Diseases and Its Role in Doxorubicin-Induced Cardiotoxicity" International Journal of Molecular Sciences 23, no. 23: 14649. https://doi.org/10.3390/ijms232314649