
Is There a Role for Immunoregulatory and Antiviral Oligonucleotides Acting in the Extracellular Space? A Review and Hypothesis

Abstract
:1. Introduction
2. Characteristics of Therapeutic Oligonucleotides
2.1. ASO
2.2. siRNA
2.3. Others
3. The Expanding Identification of Small Non-Coding RNAs
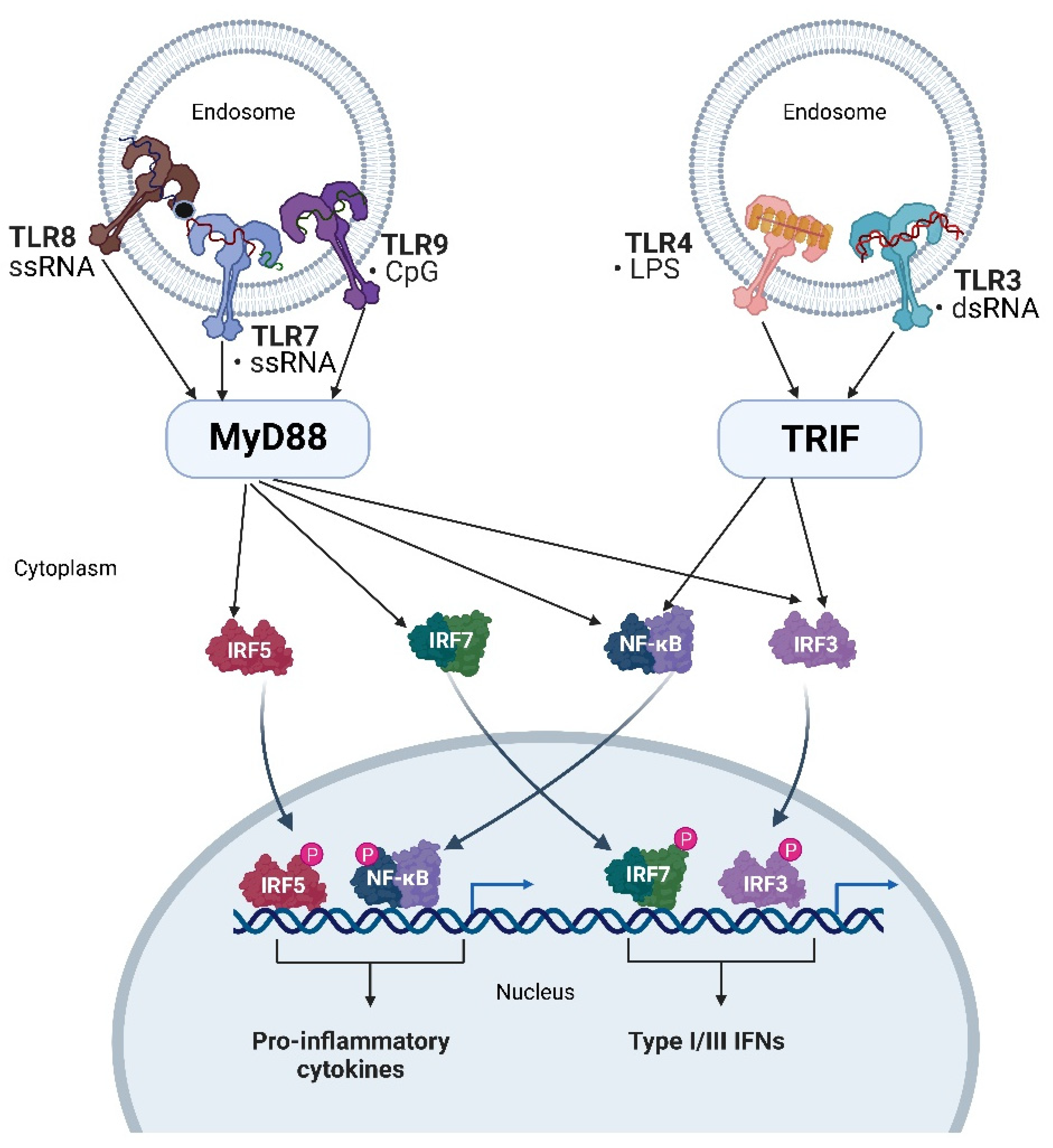
4. Endosomal Immune Receptors Recognizing Nucleic Acids
5. Discovery of SOMIE
6. ssONs Acting as Attachment/Entry Inhibitors of Viruses
7. Nucleolin Is a Binding Partner for ssONs
8. Therapeutic Approaches of ssONs Acting in the Extracellular Space
9. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vaillant, A. Oligonucleotide-Based Therapies for Chronic HBV Infection: A Primer on Biochemistry, Mechanisms and Antiviral Effects. Viruses 2022, 14, 2052. [Google Scholar] [CrossRef]
- Poux, C.; Dondalska, A.; Bergenstrahle, J.; Palsson, S.; Contreras, V.; Arasa, C.; Jarver, P.; Albert, J.; Busse, D.C.; LeGrand, R.; et al. A Single-Stranded Oligonucleotide Inhibits Toll-Like Receptor 3 Activation and Reduces Influenza A (H1N1) Infection. Front. Immunol. 2019, 10, 2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson, S.A.; Dondalska, A.; Bergenstrahle, J.; Rolfes, C.; Bjork, A.; Sedano, L.; Power, U.F.; Rameix-Welti, M.A.; Lundeberg, J.; Wahren-Herlenius, M.; et al. Single-Stranded Oligonucleotide-Mediated Inhibition of Respiratory Syncytial Virus Infection. Front. Immunol. 2020, 11, 580547. [Google Scholar] [CrossRef] [PubMed]
- Palsson, S.A.; Sekar, V.; Kutter, C.; Friedlander, M.R.; Spetz, A.L. Inhibition of Respiratory Syncytial Virus Infection by Small Non-Coding RNA Fragments. Int. J. Mol. Sci. 2022, 23, 5990. [Google Scholar] [CrossRef] [PubMed]
- Cena-Diez, R.; Singh, K.; Spetz, A.L.; Sonnerborg, A. Novel Naturally Occurring Dipeptides and Single-Stranded Oligonucleotide Act as Entry Inhibitors and Exhibit a Strong Synergistic Anti-HIV-1 Profile. Infect. Dis. Ther. 2022, 11, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, A.; Juteau, J.M.; Lu, H.; Liu, S.; Lackman-Smith, C.; Ptak, R.; Jiang, S. Phosphorothioate oligonucleotides inhibit human immunodeficiency virus type 1 fusion by blocking gp41 core formation. Antimicrob. Agents Chemother. 2006, 50, 1393–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.M.; Rojek, J.M.; Gundersen, A.; Stroher, U.; Juteau, J.M.; Vaillant, A.; Kunz, S. Inhibition of cellular entry of lymphocytic choriomeningitis virus by amphipathic DNA polymers. Virology 2008, 372, 107–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocisko, D.A.; Vaillant, A.; Lee, K.S.; Arnold, K.M.; Bertholet, N.; Race, R.E.; Olsen, E.A.; Juteau, J.M.; Caughey, B. Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob. Agents Chemother. 2006, 50, 1034–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beilstein, F.; Blanchet, M.; Vaillant, A.; Sureau, C. Nucleic Acid Polymers Are Active against Hepatitis Delta Virus Infection In Vitro. J. Virol. 2018, 92, e01416–e01417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaillant, A. REP 2139: Antiviral Mechanisms and Applications in Achieving Functional Control of HBV and HDV Infection. ACS Infect. Dis. 2019, 5, 675–687. [Google Scholar] [CrossRef]
- Boulon, R.; Blanchet, M.; Lemasson, M.; Vaillant, A.; Labonte, P. Characterization of the antiviral effects of REP 2139 on the HBV lifecycle in vitro. Antiviral. Res. 2020, 183, 104853. [Google Scholar] [CrossRef] [PubMed]
- Gait, M.J.; Agrawal, S. Introduction and History of the Chemistry of Nucleic Acids Therapeutics. Methods Mol. Biol. 2022, 2434, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Ranjith-Kumar, C.T.; Duffy, K.E.; Jordan, J.L.; Eaton-Bassiri, A.; Vaughan, R.; Hoose, S.A.; Lamb, R.J.; Sarisky, R.T.; Kao, C.C. Single-stranded oligonucleotides can inhibit cytokine production induced by human toll-like receptor 3. Mol. Cell. Biol. 2008, 28, 4507–4519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skold, A.E.; Hasan, M.; Vargas, L.; Saidi, H.; Bosquet, N.; Le Grand, R.; Smith, C.I.; Spetz, A.L. Single-stranded DNA oligonucleotides inhibit TLR3-mediated responses in human monocyte-derived dendritic cells and in vivo in cynomolgus macaques. Blood 2012, 120, 768–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarver, P.; Dondalska, A.; Poux, C.; Sandberg, A.; Bergenstrahle, J.; Skold, A.E.; Dereuddre-Bosquet, N.; Martinon, F.; Palsson, S.; Zaghloul, E.; et al. Single-Stranded Nucleic Acids Regulate TLR3/4/7 Activation through Interference with Clathrin-Mediated Endocytosis. Sci. Rep. 2018, 8, 15841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bost, J.P.; Barriga, H.; Holme, M.N.; Gallud, A.; Maugeri, M.; Gupta, D.; Lehto, T.; Valadi, H.; Esbjorner, E.K.; Stevens, M.M.; et al. Delivery of Oligonucleotide Therapeutics: Chemical Modifications, Lipid Nanoparticles, and Extracellular Vesicles. ACS Nano 2021, 15, 13993–14021. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Roehr, B. Fomivirsen approved for CMV retinitis. J. Int. Assoc. Physicians AIDS Care 1998, 4, 14–16. [Google Scholar]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2’-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998, 273, 20556–20567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R.; Group, V.I.S.i.O.N.C.T. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef] [Green Version]
- Crooke, R.M.; Graham, M.J.; Lemonidis, K.M.; Whipple, C.P.; Koo, S.; Perera, R.J. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J. Lipid. Res. 2005, 46, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; Aggarwal, S.; Topaloglu, O.; Villa, K.F.; Corbacioglu, S. Systematic review of defibrotide studies in the treatment of veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS). Bone Marrow Transpl. 2019, 54, 1951–1962. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Riches, M.L.; Kernan, N.A.; Brochstein, J.A.; Mineishi, S.; Termuhlen, A.M.; Arai, S.; Grupp, S.A.; Guinan, E.C.; Martin, P.L.; et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood 2016, 127, 1656–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, E.J.; Guo, S.; Benson, M.D.; Booten, S.; Freier, S.; Hughes, S.G.; Kim, T.W.; Jesse Kwoh, T.; Matson, J.; Norris, D.; et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid 2016, 23, 148–157. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Plante-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiro, P.A.; Rees, D.C.; Stolzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef]
- Servais, L.; Mercuri, E.; Straub, V.; Guglieri, M.; Seferian, A.M.; Scoto, M.; Leone, D.; Koenig, E.; Khan, N.; Dugar, A.; et al. Long-Term Safety and Efficacy Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A First-in-human, Multicenter, Two-Part, Open-Label, Phase 1/2 Trial. Nucleic Acid Ther. 2022, 32, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Komaki, H.; Nagata, T.; Saito, T.; Masuda, S.; Takeshita, E.; Sasaki, M.; Tachimori, H.; Nakamura, H.; Aoki, Y.; Takeda, S. Systemic administration of the antisense oligonucleotide NS-065/NCNP-01 for skipping of exon 53 in patients with Duchenne muscular dystrophy. Sci. Transl. Med. 2018, 10, eaan0713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2021, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Kuntz, N.L.; Koenig, E.; East, L.; Upadhyay, S.; Han, B.; Shieh, P.B. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: A randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve 2021, 64, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Plante-Bordeneuve, V.; Berk, J.L.; Gonzalez-Duarte, A.; Gillmore, J.D.; Low, S.C.; Sekijima, Y.; et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: A randomized clinical trial. Amyloid 2022, 1–9. [Google Scholar] [CrossRef]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosace, D.; Lopez, J.; Blanco, S. Emerging roles of novel small non-coding regulatory RNAs in immunity and cancer. RNA Biol. 2020, 17, 1196–1213. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhou, T.; Chen, Q. Exploring the expanding universe of small RNAs. Nat. Cell Biol. 2022, 24, 415–423. [Google Scholar] [CrossRef]
- Wang, H.; Huang, R.; Li, L.; Zhu, J.; Li, Z.; Peng, C.; Zhuang, X.; Lin, H.; Shi, S.; Huang, P. CPA-seq reveals small ncRNAs with methylated nucleosides and diverse termini. Cell Discov. 2021, 7, 25. [Google Scholar] [CrossRef]
- Alfonzo, J.D.; Brown, J.A.; Byers, P.H.; Cheung, V.G.; Maraia, R.J.; Ross, R.L. A call for direct sequencing of full-length RNAs to identify all modifications. Nat. Genet. 2021, 53, 1113–1116. [Google Scholar] [CrossRef]
- Zhuang, X. Spatially resolved single-cell genomics and transcriptomics by imaging. Nat. Methods 2021, 18, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Frisen, J.; Lundeberg, J. Spatially resolved transcriptomics adds a new dimension to genomics. Nat. Methods 2021, 18, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Ptashkin, R.N.; Chen, Y.; Cheng, Z.; Liu, G.; Phan, T.; Deng, X.; Zhou, J.; Lee, I.; Lee, Y.S.; et al. Respiratory Syncytial Virus Utilizes a tRNA Fragment to Suppress Antiviral Responses Through a Novel Targeting Mechanism. Mol. Ther. 2015, 23, 1622–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, M.H.; Skalsky, R.L. Roles of Non-coding RNAs During Herpesvirus Infection. Curr. Top. Microbiol. Immunol. 2018, 419, 243–280. [Google Scholar] [CrossRef] [PubMed]
- Henzinger, H.; Barth, D.A.; Klec, C.; Pichler, M. Non-Coding RNAs and SARS-Related Coronaviruses. Viruses 2020, 12, 1374. [Google Scholar] [CrossRef] [PubMed]
- Pelka, K.; Shibata, T.; Miyake, K.; Latz, E. Nucleic acid-sensing TLRs and autoimmunity: Novel insights from structural and cell biology. Immunol. Rev. 2016, 269, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Marshak-Rothstein, A. Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 2006, 6, 823–835. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Shi, J.; Liu, H.; Zhang, X.; Zhou, J.J.; Li, M.; Zhou, D.; Li, R.; Lv, J.; Wen, G.; et al. Peripheral blood non-canonical small non-coding RNAs as novel biomarkers in lung cancer. Mol. Cancer 2020, 19, 159. [Google Scholar] [CrossRef]
- Cambier, L.; de Couto, G.; Ibrahim, A.; Echavez, A.K.; Valle, J.; Liu, W.; Kreke, M.; Smith, R.R.; Marban, L.; Marban, E. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol. Med. 2017, 9, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Heard, E. Small RNAs derived from structural non-coding RNAs. Methods 2013, 63, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Glazov, E.A.; Lassmann, T.; Hayashizaki, Y.; Carninci, P.; Mattick, J.S. Small RNAs derived from snoRNAs. RNA 2009, 15, 1233–1240. [Google Scholar] [CrossRef] [Green Version]
- Gallo, S.; Kong, E.; Ferro, I.; Polacek, N. Small but Powerful: The Human Vault RNAs as Multifaceted Modulators of Pro-Survival Characteristics and Tumorigenesis. Cancers 2022, 14, 2787. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.T.; Gale, M., Jr.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Lind, N.A.; Rael, V.E.; Pestal, K.; Liu, B.; Barton, G.M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat. Rev. Immunol. 2022, 22, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Funami, K.; Seya, T.; Matsumoto, M. The clathrin-mediated endocytic pathway participates in dsRNA-induced IFN-beta production. J. Immunol. 2008, 181, 5522–5529. [Google Scholar] [CrossRef] [Green Version]
- Leonard, J.N.; Ghirlando, R.; Askins, J.; Bell, J.K.; Margulies, D.H.; Davies, D.R.; Segal, D.M. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. USA 2008, 105, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, K.; Watanabe, T.; Tokisue, T.; Tsujita, T.; Nishikawa, S.; Hasegawa, T.; Seya, T.; Matsumoto, M. Modulation of double-stranded RNA recognition by the N-terminal histidine-rich region of the human toll-like receptor 3. J. Biol. Chem. 2008, 283, 22787–22794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatematsu, M.; Nishikawa, F.; Seya, T.; Matsumoto, M. Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat. Commun. 2013, 4, 1833. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.K.; Botos, I.; Hall, P.R.; Askins, J.; Shiloach, J.; Segal, D.M.; Davies, D.R. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc. Natl. Acad. Sci. USA 2005, 102, 10976–10980. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Botos, I.; Wang, Y.; Leonard, J.N.; Shiloach, J.; Segal, D.M.; Davies, D.R. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 2008, 320, 379–381. [Google Scholar] [CrossRef] [Green Version]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [Green Version]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.M.; Kruger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Tanji, H.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Isobe, T.; Miyake, K.; Shimizu, T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 2015, 22, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kayraklioglu, N.; Horuluoglu, B.; Klinman, D.M. CpG Oligonucleotides as Vaccine Adjuvants. Methods Mol. Biol. 2021, 2197, 51–85. [Google Scholar] [CrossRef] [PubMed]
- Lebre, M.C.; van der Aar, A.M.; van Baarsen, L.; van Capel, T.M.; Schuitemaker, J.H.; Kapsenberg, M.L.; de Jong, E.C. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J. Investig. Dermatol. 2007, 127, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Huang, X.; McLeod, P.; Jiang, J.; Liu, W.; Haig, A.; Jevnikar, A.M.; Jiang, Z.; Zhang, Z.X. Toll-like receptor 3 is an endogenous sensor of cell death and a potential target for induction of long-term cardiac transplant survival. Am. J. Transplant. 2021, 21, 3268–3279. [Google Scholar] [CrossRef]
- Zhuang, C.; Chen, R.; Zheng, Z.; Lu, J.; Hong, C. Toll-Like Receptor 3 in Cardiovascular Diseases. Heart Lung Circ. 2022, 31, e93–e109. [Google Scholar] [CrossRef]
- Tatematsu, M.; Seya, T.; Matsumoto, M. Beyond dsRNA: Toll-like receptor 3 signalling in RNA-induced immune responses. Biochem. J. 2014, 458, 195–201. [Google Scholar] [CrossRef]
- Gao, D.; Ciancanelli, M.J.; Zhang, P.; Harschnitz, O.; Bondet, V.; Hasek, M.; Chen, J.; Mu, X.; Itan, Y.; Cobat, A.; et al. TLR3 controls constitutive IFN-beta antiviral immunity in human fibroblasts and cortical neurons. J. Clin. Investig. 2021, 131, e134529. [Google Scholar] [CrossRef]
- Wang, Y.; Swiecki, M.; McCartney, S.A.; Colonna, M. dsRNA sensors and plasmacytoid dendritic cells in host defense and autoimmunity. Immunol. Rev. 2011, 243, 74–90. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Y.; Herman, M.; Ciancanelli, M.J.; Perez de Diego, R.; Sancho-Shimizu, V.; Abel, L.; Casanova, J.L. TLR3 immunity to infection in mice and humans. Curr. Opin. Immunol. 2013, 25, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Hacker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariko, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavassani, K.A.; Ishii, M.; Wen, H.; Schaller, M.A.; Lincoln, P.M.; Lukacs, N.W.; Hogaboam, C.M.; Kunkel, S.L. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 2008, 205, 2609–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarante, M.K.; Watanabe, M.A. Toll-like receptor 3: Involvement with exogenous and endogenous RNA. Int. Rev. Immunol. 2010, 29, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Kindberg, E.; Vene, S.; Mickiene, A.; Lundkvist, A.; Lindquist, L.; Svensson, L. A functional Toll-like receptor 3 gene (TLR3) may be a risk factor for tick-borne encephalitis virus (TBEV) infection. J. Infect. Dis. 2011, 203, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Le Goffic, R.; Balloy, V.; Lagranderie, M.; Alexopoulou, L.; Escriou, N.; Flavell, R.; Chignard, M.; Si-Tahar, M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006, 2, e53. [Google Scholar] [CrossRef]
- Wang, Q.; Miller, D.J.; Bowman, E.R.; Nagarkar, D.R.; Schneider, D.; Zhao, Y.; Linn, M.J.; Goldsmith, A.M.; Bentley, J.K.; Sajjan, U.S.; et al. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 2011, 7, e1002070. [Google Scholar] [CrossRef]
- Murray, L.A.; Knight, D.A.; McAlonan, L.; Argentieri, R.; Joshi, A.; Shaheen, F.; Cunningham, M.; Alexopolou, L.; Flavell, R.A.; Sarisky, R.T.; et al. Deleterious role of TLR3 during hyperoxia-induced acute lung injury. Am. J. Respir. Crit. Care Med. 2008, 178, 1227–1237. [Google Scholar] [CrossRef]
- Citores, M.J.; Banos, I.; Noblejas, A.; Rosado, S.; Castejon, R.; Cuervas-Mons, V. Toll-like receptor 3 L412F polymorphism may protect against acute graft rejection in adult patients undergoing liver transplantation for hepatitis C-related cirrhosis. Transplant. Proc. 2011, 43, 2224–2226. [Google Scholar] [CrossRef] [PubMed]
- Strodthoff, D.; Ma, Z.; Wirstrom, T.; Strawbridge, R.J.; Ketelhuth, D.F.; Engel, D.; Clarke, R.; Falkmer, S.; Hamsten, A.; Hansson, G.K.; et al. Toll-Like Receptor 3 Influences Glucose Homeostasis and beta-Cell Insulin Secretion. Diabetes 2015, 64, 3425–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentano, F.; Schorr, O.; Gay, R.E.; Gay, S.; Kyburz, D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum. 2005, 52, 2656–2665. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.; Dieudonne, A.; Ryffel, B.; Vilain, E.; Si-Tahar, M.; Pichavant, M.; Lassalle, P.; Trottein, F.; Gosset, P. Double-stranded RNA exacerbates pulmonary allergic reaction through TLR3: Implication of airway epithelium and dendritic cells. J. Immunol. 2010, 185, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, A.I.; Spathis, A.; Kostikas, K.; Karakitsos, P.; Papiris, S.; Rossios, C. The role of endosomal toll-like receptors in asthma. Eur. J. Pharmacol. 2017, 808, 14–20. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Ku, C.L.; Lim, H.K.; Hsia, S.H.; Lin, J.J.; Lo, C.C.; Ding, J.Y.; Kuo, R.L.; Casanova, J.L.; Zhang, S.Y.; et al. Life-Threatening Enterovirus 71 Encephalitis in Unrelated Children with Autosomal Dominant TLR3 Deficiency. J. Clin. Immunol. 2022, 42, 606–617. [Google Scholar] [CrossRef]
- Partanen, T.; Chen, J.; Lehtonen, J.; Kuismin, O.; Rusanen, H.; Vapalahti, O.; Vaheri, A.; Anttila, V.J.; Bode, M.; Hautala, N.; et al. Heterozygous TLR3 Mutation in Patients with Hantavirus Encephalitis. J. Clin. Immunol. 2020, 40, 1156–1162. [Google Scholar] [CrossRef]
- Lim, H.K.; Huang, S.X.L.; Chen, J.; Kerner, G.; Gilliaux, O.; Bastard, P.; Dobbs, K.; Hernandez, N.; Goudin, N.; Hasek, M.L.; et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 2019, 216, 2038–2056. [Google Scholar] [CrossRef]
- Krieg, A.M.; Vollmer, J. Toll-like receptors 7, 8, and 9: Linking innate immunity to autoimmunity. Immunol. Rev. 2007, 220, 251–269. [Google Scholar] [CrossRef]
- Klinman, D.M.; Tross, D.; Klaschik, S.; Shirota, H.; Sato, T. Therapeutic applications and mechanisms underlying the activity of immunosuppressive oligonucleotides. Ann. N. Y. Acad. Sci. 2009, 1175, 80–88. [Google Scholar] [CrossRef]
- Latz, E.; Verma, A.; Visintin, A.; Gong, M.; Sirois, C.M.; Klein, D.C.; Monks, B.G.; McKnight, C.J.; Lamphier, M.S.; Duprex, W.P.; et al. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat. Immunol. 2007, 8, 772–779. [Google Scholar] [CrossRef]
- Wagner, H. The sweetness of the DNA backbone drives Toll-like receptor 9. Curr. Opin. Immunol. 2008, 20, 396–400. [Google Scholar] [CrossRef]
- Kuznik, A.; Panter, G.; Jerala, R. Recognition of nucleic acids by Toll-like receptors and development of immunomodulatory drugs. Curr. Med. Chem. 2010, 17, 1899–1914. [Google Scholar] [CrossRef]
- Cossart, P.; Helenius, A. Endocytosis of viruses and bacteria. Cold Spring Harb. Perspect. Biol. 2014, 6, a016972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderlinden, E.; Naesens, L. Emerging antiviral strategies to interfere with influenza virus entry. Med. Res. Rev. 2014, 34, 301–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco-Garcia, A.S.; Garrido-Martin, E.M.; Rupani, H.; Lau, L.C.K.; Martinez-Nunez, R.T.; Howarth, P.H.; Sanchez-Elsner, T. Small RNA Species and microRNA Profiles are Altered in Severe Asthma Nanovesicles from Broncho Alveolar Lavage and Associate with Impaired Lung Function and Inflammation. Noncoding RNA 2019, 5, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Gorospe, M. RNA-binding protein nucleolin in disease. RNA Biol. 2012, 9, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Hovanessian, A.G.; Soundaramourty, C.; El Khoury, D.; Nondier, I.; Svab, J.; Krust, B. Surface expressed nucleolin is constantly induced in tumor cells to mediate calcium-dependent ligand internalization. PLoS ONE 2010, 5, e15787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Liang, C.; Li, F.; Wang, L.; Wu, X.; Lu, A.; Xiao, G.; Zhang, G. The Rules and Functions of Nucleocytoplasmic Shuttling Proteins. Int. J. Mol. Sci. 2018, 19, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, N.; Ding, Y.; Zhuo, W.; He, T.; Fu, Z.; Chen, Y.; Song, X.; Fu, Y.; Luo, Y. The nuclear translocation of endostatin is mediated by its receptor nucleolin in endothelial cells. Angiogenesis 2012, 15, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Matsuda, T.; Washizaki, A.; Murakami, H.; Yamamoto, T.; Yoshioka, Y. Elucidation of the role of nucleolin as a cell surface receptor for nucleic acid-based adjuvants. NPJ Vaccines 2022, 7, 115. [Google Scholar] [CrossRef]
- Tayyari, F.; Marchant, D.; Moraes, T.J.; Duan, W.; Mastrangelo, P.; Hegele, R.G. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 2011, 17, 1132–1135. [Google Scholar] [CrossRef]
- Mastrangelo, P.; Chin, A.A.; Tan, S.; Jeon, A.H.; Ackerley, C.A.; Siu, K.K.; Lee, J.E.; Hegele, R.G. Identification of RSV Fusion Protein Interaction Domains on the Virus Receptor, Nucleolin. Viruses 2021, 13, 261. [Google Scholar] [CrossRef]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef]
- Song, J.; Quan, R.; Wang, D.; Liu, J. Seneca Valley Virus 3C(pro) Mediates Cleavage and Redistribution of Nucleolin to Facilitate Viral Replication. Microbiol. Spectr. 2022, 10, e0030422. [Google Scholar] [CrossRef]
- Tonello, F.; Massimino, M.L.; Peggion, C. Nucleolin: A cell portal for viruses, bacteria, and toxins. Cell Mol. Life Sci. 2022, 79, 271. [Google Scholar] [CrossRef]
- Mastrangelo, P.; Norris, M.J.; Duan, W.; Barrett, E.G.; Moraes, T.J.; Hegele, R.G. Targeting Host Cell Surface Nucleolin for RSV Therapy: Challenges and Opportunities. Vaccines 2017, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, P.; Hegele, R.G. The RSV fusion receptor: Not what everyone expected it to be. Microbes Infect. 2012, 14, 1205–1210. [Google Scholar] [CrossRef]
- Anderson, C.S.; Chirkova, T.; Slaunwhite, C.G.; Qiu, X.; Walsh, E.E.; Anderson, L.J.; Mariani, T.J. CX3CR1 Engagement by Respiratory Syncytial Virus Leads to Induction of Nucleolin and Dysregulation of Cilia-related Genes. J. Virol. 2021, 95, e00095-21. [Google Scholar] [CrossRef]
- Griffiths, C.D.; Bilawchuk, L.M.; McDonough, J.E.; Jamieson, K.C.; Elawar, F.; Cen, Y.; Duan, W.; Lin, C.; Song, H.; Casanova, J.L.; et al. IGF1R is an entry receptor for respiratory syncytial virus. Nature 2020, 583, 615–619. [Google Scholar] [CrossRef]
- Dondalska, A.; Ronnberg, E.; Ma, H.; Palsson, S.A.; Magnusdottir, E.; Gao, T.; Adam, L.; Lerner, E.A.; Nilsson, G.; Lagerstrom, M.; et al. Amelioration of Compound 48/80-Mediated Itch and LL-37-Induced Inflammation by a Single-Stranded Oligonucleotide. Front. Immunol. 2020, 11, 559589. [Google Scholar] [CrossRef] [PubMed]
- Gokirmak, T.; Nikan, M.; Wiechmann, S.; Prakash, T.P.; Tanowitz, M.; Seth, P.P. Overcoming the challenges of tissue delivery for oligonucleotide therapeutics. Trends Pharmacol. Sci. 2021, 42, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, T.; Morales-Medina, J.C.; Palmieri, B. Phosphorothioate oligonucleotides: Effectiveness and toxicity. Curr. Drug Targets 2014, 15, 663–673. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trade Name (Name), Company | Class | Chemistry | Indication, (Target), Organ | FDA/EMA Approval Year | Comment |
|---|---|---|---|---|---|
| Vitravene (fomivirsen) Ionis Pharma Novartis | ASO | 21-mer PS DNA | CMV renitis, (viral IE2 mRNA), eye | 1998 | First approved nucleic acid drug [18]. Withdrawn from use due to reduced clinical need. Mechanism unclear. |
| Macugen, (pegaptanib) NeXstar Pharma Eyetech/Pfizer | Aptamer | 28-mer 2′-F/2′-OMe/pegylated RNA [19,20] | Age-related macular degeneration (VEGF-165), eye | 2004 | Anti-angiogenic, intravitreal injection. |
| Kynamro (mipomersen) Ionis Pharma, Genzyme Kastle Tx | Gapmer ASO | 20-mer PS 2′-MOE [21] | Homozygous familial hypercholesterolaemia, (APOB mRNA), liver | 2013 | RNase H-mediated cleavage of apolipoprotein B mRNA. Subcutaneous (SC) injection. |
| Defitelio (defibrotide), Jazz Pharma | Mix of DNA isolated from porcine mucosa | Mix of PO- ssDNA and dsDNA | Hepatic veno-occlusive disease [22], (NA), liver | 2016 | Sequence-independent mechanism of action. Intravenous injection (IV) [23]. |
| Exondys 51 (eteplirsen), Sarepta Tx | ASO | 30-mer PMO | Duchenne muscular dystrophy, (DMD exon 51), skeletal muscle | 2016 | Steric block, splice-switching, [24] IV injection. |
| Spinraza (nusinersen) Ionis Pharma Biogen | ASO | 18-mer PS 2′-MOE | Spinal muscular atrophy, (SMN2 exon 7), CNS | 2016 | Steric block, splice-switching [25], Intrathecal injection. |
| Onpattro (patisiran) Alnylam Pharma | siRNA | 19+2¤-mer 2′-OMe, ds [26] | Hereditary transthyretin-mediated amyloidosis, (TTR), liver | 2018 | Lipid nanoparticle formulation, IV injection. |
| Tegsedi (inotersen) Inonis Pharma Akcea Pharma | Gapmer ASO | 20-mer PS 2′-MOE [27] | Hereditary transthyretin amyloidosis, (TTR), liver | 2018 | RNaseH mechanism of action, leading to reductions in TTR protein [28], SC injection. |
| Waylivra (volanesorsen) Ionis Pharma Akcea Pharma | Gapmer ASO | 20-mer PS 2′-MOE [29] | Familial chylomicronaemia syndrome, (APOC3), liver | 2019 | Only approved by EMA not FDA. RNaseH mechanism of action, leading to reductions in apoC3 proteins, SC injection. |
| Givlaari (givosiran) Alnylam Pharma | siRNA | 21/23-mer With partial PS, 2′-F, 2′-OMe, ds [30] | Acute hepatic porphyria (ALAS1), liver | 2019 | GalNAc conjugate to target hepatocytes, SC injection. |
| Vyondys 53 (golodirsen) Sarepta Tx | ASO | 25-mer PMO | Duchenne muscular dystrophy, (DMD exon 53), skeletal muscle | 2019 | Splice-switching [31], IV injection. |
| Viltepso (viltolarsen) NS Pharma | ASO | 21-mer PMO | Duchenne muscular dystrophy, (DMD exon 53), skeletal muscle | 2020 | Splice-switching [32], IV injection. |
| Oxlumo (lumasiran) Alnylam Pharma | siRNA | 21/23-mer With partial PS, 2′-F, 2′-OMe, ds [33] | Primary hyperoxaluria type 1 (HAO1), liver | 2020 | GalNAc conjugate to target hepatocytes, SC injection. |
| Leqvio (inclisiran) Alnylam Pharma Novartis | siRNA | 21/23-mer With partial PS, 2′-F, 2′-OMe, ds * | Hypercholesterolemia (PCSK9), liver | 2021 | GalNAc conjugate to target hepatocytes, SC injection. |
| Amondys 45 (casimersen) Sarepta Tx | ASO | 22-mer PMO | Duchenne muscular dystrophy, (DMD exon 45), skeletal muscle | 2021 | Splice-switching, [34] IV injection. |
| Amvuttra (vutrisiran) Alnylam Pharma | siRNA | 21/23-mer With partial PS, 2′-F, 2′-OMe, ds ^ | Hereditary transthyretin amyloidosis [35] (TTR), liver | 2022 | GalNAc conjugate to target hepatocytes, SC injection. |
| sncRNA | Length | Comment on Mechanism and/or Function | Ref. |
|---|---|---|---|
| siRNA | 20–27 nt | Base pairing and gene silencing with AGO. | [48] |
| miRNA | 21–23 nt | Base pairing and gene silencing with AGO. | [48] |
| piRNA | 21–35 nt | Base pairing and gene silencing with PIWI. | [49] |
| tsRNA -tRF-1 -tRF-3 -tRF-5 -3′tRNA halves -5′tRNA halves | 13–40 nt 13–30 nt 13–30 nt 13–30 nt 30–40 nt 30–40 nt | Gene silencing not always sequence dependent. Tumor suppression, T cell inhibition, affect virus replication and influence stress responses. Transfer RNAs are characterized by their typical cloverleaf structure which can be processed into abundant fragments. | [37] |
| rsRNA -several names of identified fragments. | Multiple lengths | Shorter 19–24 nt involved in gene silencing with AGO. Longer 74–130 nt function unclear. Often derived from 45S, 5S, and 28S rRNAs in PBMCs with lengths of 15–42. | [37,50] |
| ysRNA | Multiple lengths often 26–40 nt but also 83–112 nt | Overexpression of a 57 nt increased IL-10 production and administration in vivo in rats conferred cardioprotection. Sequencing in human PBMCs revealed abundant ysRNAs of approx. 26–40 nt. Often derived from YRNA-RNY4 and YRNA-RNY1 in PBMCs | [50,51] |
| snsRNA | Approx. 16–40 nt | SnRNA is as a family of highly conserved ncRNAs located in the nucleus and associated with Sm ribonucleoproteins and other specific proteins, to form small nuclear ribonucleoproteins. The function of fragments is largely unknown. | [39,52] |
| snosRNA | Often 20–24 nt but also 17–19 nt and 27–33 nt | Some were shown to be similar to miRNA and can use AGO for gene silencing. | [52,53] |
| vtsRNA | Full-length vRNA is approx. 100 nt and can be processed into approx. 23 nt | RNA components of Vault ribonucleoprotein particles, which are located in the cytoplasm. Control of apoptosis and autophagy, lysosome biogenesis, and function in cancer cells. | [54] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dondalska, A.; Axberg Pålsson, S.; Spetz, A.-L. Is There a Role for Immunoregulatory and Antiviral Oligonucleotides Acting in the Extracellular Space? A Review and Hypothesis. Int. J. Mol. Sci. 2022, 23, 14593. https://doi.org/10.3390/ijms232314593
Dondalska A, Axberg Pålsson S, Spetz A-L. Is There a Role for Immunoregulatory and Antiviral Oligonucleotides Acting in the Extracellular Space? A Review and Hypothesis. International Journal of Molecular Sciences. 2022; 23(23):14593. https://doi.org/10.3390/ijms232314593
Chicago/Turabian StyleDondalska, Aleksandra, Sandra Axberg Pålsson, and Anna-Lena Spetz. 2022. "Is There a Role for Immunoregulatory and Antiviral Oligonucleotides Acting in the Extracellular Space? A Review and Hypothesis" International Journal of Molecular Sciences 23, no. 23: 14593. https://doi.org/10.3390/ijms232314593