
Probiogenomic In-Silico Analysis and Safety Assessment of Lactiplantibacillus plantarum DJF10 Strain Isolated from Korean Raw Milk
 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
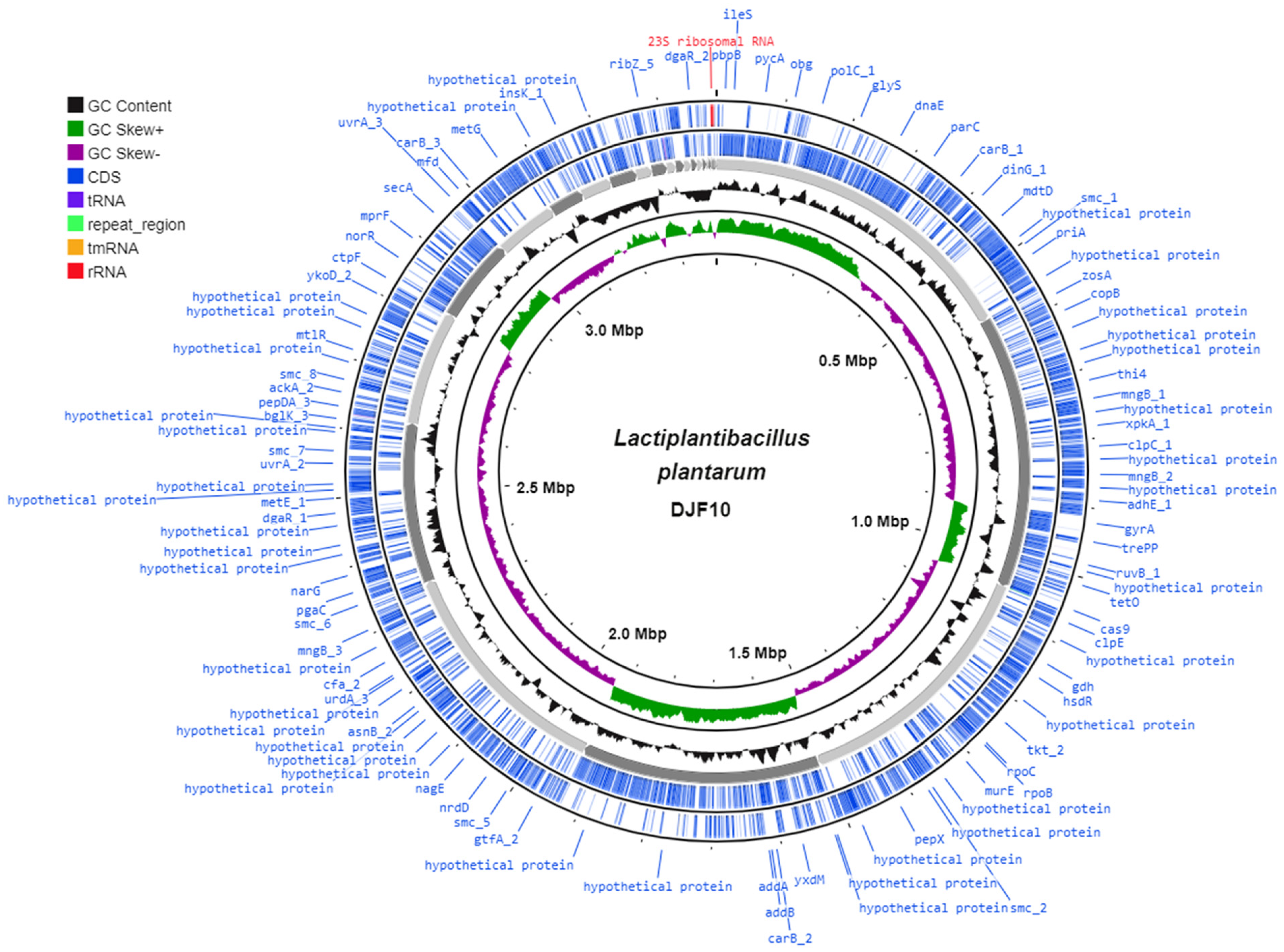
2.1. Genome Characteristics of L. plantarum DJF10
2.2. Species Confirmation
2.3. Annotation
2.3.1. Functional Prediction
2.3.2. Probiotic Properties
2.4. Carbohydrate-Active Enzymes (CAZymes)
2.5. Mobile Genetic Elements
2.5.1. Insertion Sequences
2.5.2. Island Viewer
2.5.3. CRISPR–Cas
2.5.4. Prophages
2.6. Safety-Associated Genes
2.6.1. Antimicrobial Resistance (AMR) Genes
2.6.2. Virulence Factors
2.6.3. Toxins, Biogenic Amines and Undesirable Genes
2.7. Bacteriocin-Encoding Genes
3. Materials and Methods
3.1. Strain Information
3.2. DNA Extraction, Whole Genome Sequencing, Assembly and Annotation
3.3. Genome-Based Identification
3.4. Annotation and Functional Prediction
3.5. Genome Instability
3.6. Safety Assessment
3.7. Secondary Metabolites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sánchez, B.; Delgado, S.; Blanco-Míguez, A.; Lourenço, A.; Gueimonde, M.; Margolles, A. Probiotics, gut microbiota, and their influence on host health and disease. Mol. Nutr. Food Res. 2017, 61, 1600240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pot, B.; Felis, G.E.; De Bruyne, K.; Tsakalidou, E.; Papadimitriou, K.; Leisner, J.; Vanadmme, P. The genus Lactobacillus. In Lactic Acid Bacteria: Biodiversity and Taxonomy; Holzapfel, W.H., Wood, B.J.B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 249–353. [Google Scholar] [CrossRef]
- Leuschner, R.G.K.; Robinson, T.P.; Hugas, M.; Cocconcelli, P.S.; Richard-Forget, F.; Klein, G.; Licht, T.R.; Nguyen-The, C.; Querol, A.; Richardson, M.; et al. Qualified presumption of safety (QPS): A generic risk assessment approach for biological agents notified to the European Food Safety Authority (EFSA). Trends Food Sci. Technol. 2010, 21, 425–435. [Google Scholar] [CrossRef]
- Laulund, S.; Wind, A.; Derkx, P.M.F.; Zuliani, V. Regulatory and Safety Requirements for Food Cultures. Microorganisms 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gareau, M.G.; Sherman, P.M.; Walker, W.A. Probiotics and the gut microbiota in intestinal health and disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Melo Pereira, G.V.; de Oliveira Coelho, B.; Júnior, A.I.M.; Thomaz-Soccol, V.; Soccol, C.R. How to select a probiotic? A review and update of methods and criteria. Biotechnol. Adv. 2018, 36, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Daliri, E.B.M.; Lee, B.H.; Oh, D.H. Safety of Probiotics in Health and Disease. In The Role of Functional Food Security in Global Health; Academic Press: Cambridge, MA, USA, 2019; pp. 603–622. [Google Scholar] [CrossRef]
- Beck, B.R.; Park, G.S.; Lee, Y.H.; Im, S.; Jeong, D.Y.; Kang, J. Whole genome analysis of Lactobacillus plantarum strains isolated from kimchi and determination of probiotic properties to treat mucosal infections by Candida albicans and gardnerella vaginalis. Front. Microbiol. 2019, 10, 433. [Google Scholar] [CrossRef]
- Arellano, K.; Vazquez, J.; Park, H.; Lim, J.; Ji, Y.; Kang, H.J.; Cho, D.; Jeong, H.W.; Holzapfel, W.H. Safety Evaluation and Whole-Genome Annotation of Lactobacillus plantarum Strains from Different Sources with Special Focus on Isolates from Green Tea. Probiotics Antimicrob. Proteins 2020, 12, 1057–1070. [Google Scholar] [CrossRef]
- Vemuri, R.; Gundamaraju, R.; Eri, R. Role of Lactic Acid Probiotic Bacteria in IBD. Curr. Pharm. Des. 2017, 23, 2352–2355. [Google Scholar] [CrossRef]
- Borase, H.; Dwivedi, M.K.; Krishnamurthy, R.; Patil, S. Probiotics: Health safety considerations. In Probiotics in the Prevention and Management of Human Diseases; Academic Press: Cambridge, MA, USA, 2022; pp. 449–463. [Google Scholar]
- Peng, X.; Ed-Dra, A.; Yue, M. Whole genome sequencing for the risk assessment of probiotic lactic acid bacteria. Crit. Rev. Food Sci. Nutr. 2022, 1–19. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Q.; Lu, B.; Shen, H.; Liu, S.; Shi, Y.; Leptihn, S.; Li, H.; Wei, J.; Liu, C.; et al. Whole-genome analysis of probiotic product isolates reveals the presence of genes related to antimicrobial resistance, virulence factors, and toxic metabolites, posing potential health risks. BMC Genom. 2021, 22, 210. [Google Scholar] [CrossRef]
- Syrokou, M.K.; Paramithiotis, S.; Drosinos, E.H.; Bosnea, L.; Mataragas, M. A Comparative Genomic and Safety Assessment of Six Lactiplantibacillus plantarum subsp. argentoratensis Strains Isolated from Spontaneously Fermented Greek Wheat Sourdoughs for Potential Biotechnological Application. Int. J. Mol. Sci. 2022, 23, 2487. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Yang, S.M.; Kim, D.; Kim, H.Y. Complete Genome Sequencing and Comparative Genomics of Three Potential Probiotic Strains, Lacticaseibacillus casei FBL6, Lacticaseibacillus chiayiensis FBL7, and Lacticaseibacillus zeae FBL8. Front. Microbiol. 2022, 12, 794315. [Google Scholar] [CrossRef]
- Jia, F.F.; Zhang, L.J.; Pang, X.H.; Gu, X.X.; Abdelazez, A.; Liang, Y.; Sun, S.R.; Meng, X.C. Complete genome sequence of bacteriocin-producing Lactobacillus plantarum KLDS1.0391, a probiotic strain with gastrointestinal tract resistance and adhesion to the intestinal epithelial cells. Genomics 2017, 109, 432–437. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, Y.; Sun, M.; Zhang, H.; Mu, G.; Tuo, Y. Physiological function analysis of Lactobacillus plantarum Y44 based on genotypic and phenotypic characteristics. J. Dairy Sci. 2020, 103, 5916–5930. [Google Scholar] [CrossRef] [PubMed]
- Surve, S.; Shinde, D.B.; Kulkarni, R. Isolation, characterization and comparative genomics of potentially probiotic Lactiplantibacillus plantarum strains from Indian foods. Sci. Rep. 2022, 12, 1940. [Google Scholar] [CrossRef]
- Rocha, C.; Danchin, A. Base composition bias might result from competition for. Trends Genet. 2002, 18, 291–294. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A Web Server for Prokaryotic Species Circumscription Based on Pairwise Genome Comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Kim, E.; Chang, H.C.; Kim, H.Y. Complete Genome Sequence of Lactobacillus plantarum EM, A Putative Probiotic Strain with the Cholesterol-Lowering Effect and Antimicrobial Activity. Curr. Microbiol. 2020, 77, 1871–1882. [Google Scholar] [CrossRef]
- Li, O.; Zhang, H.; Wang, W.; Liang, Y.; Chen, W.; Ud Din, A.; Li, L.; Zhou, Y. Complete genome sequence and probiotic properties of Lactococcus petauri LZys1 isolated from healthy human gut. J. Med. Microbiol. 2021, 70, 001397. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Wang, R.; Gong, F.M.; Liu, X.F.; Zheng, H.J.; Luo, Y.Y.; Li, X.R. Complete genome sequences and comparative genome analysis of Lactobacillus plantarum strain 5-2 isolated from fermented soybean. Genomics 2015, 106, 404–411. [Google Scholar] [CrossRef]
- Song, H.; Kim, J.; Guk, J.H.; Kim, W.H.; Nam, H.; Suh, J.G.; Seong, J.K.; Cho, S. Metagenomic Analysis of the Gut Microbiota of Wild Mice, a Newly Identified Reservoir of Campylobacter. Front. Cell. Infect. Microbiol. 2021, 10, 596149. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, S.; Li, H.; Zhu, J.; Liu, Z.; Hu, X.; Yi, J. Assessments of Probiotic Potentials of Lactiplantibacillus plantarum Strains Isolated from Chinese Traditional Fermented Food: Phenotypic and Genomic Analysis. Front. Microbiol. 2022, 13, 1–10. [Google Scholar] [CrossRef]
- Zhang, W.; Ji, H.; Zhang, D.; Liu, H.; Wang, S.; Wang, J.; Wang, Y. Complete Genome Sequencing of Lactobacillus plantarum ZLP001, a Potential Probiotic That Enhances Intestinal Epithelial Barrier Function and Defense Against Pathogens in Pigs. Front. Physiol. 2018, 9, 1689. [Google Scholar] [CrossRef] [PubMed]
- Kleerebezem, M.; Boekhorst, J.; van Kranenburg, R.; Molenaar, D.; Kuipers, O.P.; Leer, R.; Tarchini, R.; Peters, S.A.; Sandbrink, H.M.; Fiers, M.W.E.J.; et al. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. USA 2003, 100, 1990–1995. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.Y.; Liao, Y.C.; Lin, S.H.; Lin, J.S.; Lee, C.C.; Watanabe, K. Safety Assessment of Lactiplantibacillus plantarum TWK10 Based on Whole-Genome Sequencing, Phenotypic, and Oral Toxicity Analysis. Microorganisms 2022, 10, 784. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P. Pathogenicity and other genomic islands. In Brenners Encyclopedia of Genetics, 2nd ed.; Academic Press: San Diego, CA, USA, 2013; pp. 240–242. [Google Scholar] [CrossRef]
- Pei, Z.; Sadiq, F.A.; Han, X.; Zhao, J.; Zhang, H.; Ross, R.P.; Lu, W.; Chen, W. Identification, characterization, and phylogenetic analysis of eight new inducible prophages in Lactobacillus. Virus Res. 2020, 286, 198003. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; Van Der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef]
- Yetiman, A.E.; Keskin, A.; Darendeli, B.N.; Kotil, S.E.; Ortakci, F.; Dogan, M. Characterization of genomic, physiological, and probiotic features Lactiplantibacillus plantarum DY46 strain isolated from traditional lactic acid fermented shalgam beverage. Food Biosci. 2022, 46, 101499. [Google Scholar] [CrossRef]
- Campedelli, I.; Mathur, H.; Salvetti, E.; Clarke, S.; Rea, M.C.; Torriani, S.; Ross, R.P.; Hill, C.; O’Toole, P.W. Genus-wide assessment of antibiotic resistance in Lactobacillus spp. Appl. Environ. Microbiol. 2019, 85, e01738-18. [Google Scholar] [CrossRef] [Green Version]
- Chokesajjawatee, N.; Santiyanont, P.; Chantarasakha, K.; Kocharin, K.; Thammarongtham, C.; Lertampaiporn, S.; Vorapreeda, T.; Srisuk, T.; Wongsurawat, T.; Jenjaroenpun, P.; et al. Safety Assessment of a Nham Starter Culture Lactobacillus Plantarum BCC9546 via Whole-Genome Analysis. Sci. Rep. 2020, 10, 10241. [Google Scholar] [CrossRef]
- Ho Sui, S.J.; Fedynak, A.; Hsiao, W.W.L.; Langille, M.G.I.; Brinkman, F.S.L. The Association of Virulence Factors with Genomic Islands. PLoS ONE 2009, 4, e8094. [Google Scholar] [CrossRef] [PubMed]
- Zafar, H.; Saier, M.H. Comparative genomics of the transport proteins of ten lactobacillus strains. Genes 2020, 11, 1234. [Google Scholar] [CrossRef]
- De Almeida, M.O.; Carvalho, R.; Aburjaile, F.F.; Miranda, F.M.; Cerqueira, J.C.; Brenig, B.; Ghosh, P.; Ramos, R.; Kato, R.B.; de Castro Soares, S.; et al. Characterization of the first vaginal Lactobacillus crispatus genomes isolated in Brazil. PeerJ 2021, 9, e11079. [Google Scholar] [CrossRef] [PubMed]
- Bianchetti, D.G.A.M.; Amelio, G.S.; Lava, S.A.G.; Bianchetti, M.G.; Simonetti, G.D.; Agostoni, C.; Fossali, E.F.; Milani, G.P. D-lactic acidosis in humans: Systematic literature review. Pediatr. Nephrol. 2017, 33, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder-Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Tenea, G.N.; Ortega, C. Genome Characterization of Lactiplantibacillus plantarum Strain UTNGt2 Originated from Theobroma grandiflorum (White Cacao) of Ecuadorian Amazon: Antimicrobial Peptides from Safety to Potential Applications. Antibiotics 2021, 10, 383. [Google Scholar] [CrossRef]
- Diep, D.B.; Straume, D.; Kjos, M.; Torres, C.; Nes, I.F. An overview of the mosaic bacteriocin pln loci from Lactobacillus plantarum. Peptides 2009, 30, 1562–1574. [Google Scholar] [CrossRef]
- Seemann, T. Shovill: Faster SPAdes Assembly of Illumina Reads. 2017. Available online: https://github.com/tseemann/shovill (accessed on 17 August 2022).
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, A.J.; De Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Value |
|---|---|
| Genome size (bp) | 3,385,113 |
| GC content (%) | 44.3 |
| Number of contigs | 29 |
| N50 (bp) | 418,773 |
| L50 | 4 |
| Plasmids | 0 |
| CDS | 3168 |
| Total RNA’s | 68 (60 tRNA + 7 rRNA + 1 tmRNA) |
| Total genes | 3235 |
| Protein-coding genes | 3168 |
| Genes assigned to COGs | 2855 |
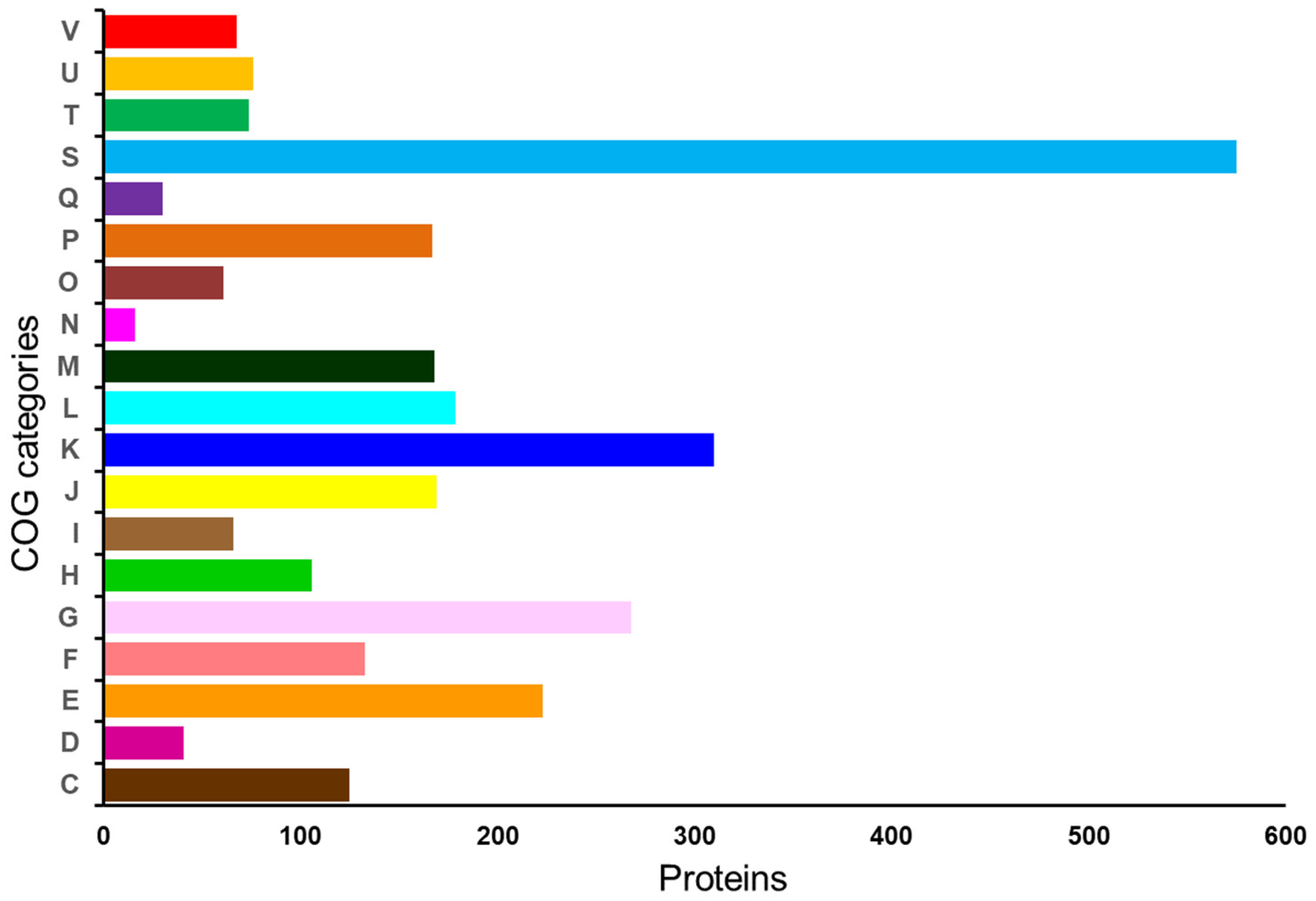
| Description | Value | Percent |
|---|---|---|
| Cofactors, Vitamins, Prosthetic Groups, Pigments | 106 | 9.5 |
| Cell Wall and Capsule | 52 | 4.6 |
| Virulence, Disease and Defense | 41 | 3.7 |
| Potassium metabolism | 6 | 0.5 |
| Miscellaneous | 14 | 1.3 |
| Phages, Prophages, Transposable elements, Plasmids | 9 | 0.8 |
| Membrane Transport | 35 | 3.1 |
| Iron acquisition and metabolism | 5 | 0.4 |
| RNA Metabolism | 38 | 3.4 |
| Nucleosides and Nucleotides | 91 | 8.1 |
| Protein Metabolism | 127 | 11.3 |
| Cell Division and Cell Cycle | 4 | 0.4 |
| Regulation and Cell signaling | 16 | 1.4 |
| Secondary Metabolism | 4 | 0.4 |
| DNA Metabolism | 63 | 5.6 |
| Fatty Acids, Lipids, and Isoprenoids | 34 | 3.0 |
| Nitrogen Metabolism | 8 | 0.7 |
| Dormancy and Sporulation | 6 | 0.5 |
| Respiration | 16 | 1.4 |
| Stress Response | 20 | 1.8 |
| Metabolism of Aromatic Compounds | 8 | 0.7 |
| Amino Acids and Derivatives | 175 | 15.6 |
| Sulfur Metabolism | 4 | 0.4 |
| Phosphorus Metabolism | 7 | 0.6 |
| Carbohydrates | 230 | 20.55 |
| KO Number | Functional Category | Gene Number | Proportion (%) |
|---|---|---|---|
| 09101 | Carbohydrate metabolism | 226 | 13.59 |
| 09102 | Energy metabolism | 37 | 2.22 |
| 09103 | Lipid metabolism | 39 | 2.35 |
| 09104 | Nucleotide metabolism | 68 | 4.09 |
| 09105 | Amino acid metabolism | 97 | 5.83 |
| 09106 | Metabolism of other amino acids | 20 | 1.20 |
| 09107 | Glycan biosynthesis and metabolism | 37 | 2.22 |
| 09108 | Metabolism of cofactors and vitamins | 65 | 3.91 |
| 09109 | Metabolism of terpenoids and polyketides | 10 | 0.60 |
| 09110 | Biosynthesis of secondary metabolites | 5 | 0.30 |
| 09111 | Xenobiotics biodegradation and metabolism | 8 | 0.48 |
| 09120 | Genetic information processing | 161 | 9.68 |
| 09130 | Environmental information processing | 164 | 9.86 |
| 09140 | Cellular processes | 11 | 0.66 |
| 09150 | Organismal systems | 8 | 0.48 |
| 09160 | Human diseases | 3 | 0.18 |
| 09181 | Protein families: metabolism | 39 | 2.35 |
| 09182 | Protein families: genetic information processing | 229 | 13.77 |
| 09183 | Protein families: signaling and cellular processes | 184 | 11.06 |
| 09191 | Unclassified: metabolism | 110 | 6.61 |
| Gene | Function | Gene Nos. |
|---|---|---|
| Temperature | ||
| Heat stress | ||
| htpX | heat shock protein HtpX | 1 |
| hrcA | heat-inducible transcriptional repressor | 1 |
| hslO | molecular chaperone Hsp33 | 1 |
| hslU | ATP-dependent HslUV protease ATP-binding subunit HslU | 1 |
| HSP20 | HSP20 family protein | 3 |
| dnaK | HSPA9; molecular chaperone DnaK | 1 |
| dnaJ | molecular chaperone DnaJ | 1 |
| ctsR | transcriptional regulator of stress and heat shock response | 1 |
| grpE | molecular chaperone GrpE | 1 |
| groEL | HSPD1; chaperonin GroEL | 1 |
| groES | HSPE1; chaperonin GroES | 1 |
| lon | Lon-like protease | 1 |
| clpB | ATP-dependent Clp protease ATP-binding subunit ClpB | 1 |
| clpC | ATP-dependent Clp protease ATP-binding subunit ClpC | 1 |
| clpE | ATP-dependent Clp protease ATP-binding subunit ClpE | 1 |
| clpL | ATP-dependent Clp protease ATP-binding subunit ClpL | 1 |
| clpX | ATP-dependent Clp protease ATP-binding subunit ClpX | 1 |
| clpP | ATP-dependent Clp protease, protease subunit | 1 |
| hslV | ATP-dependent HslUV protease, peptidase subunit HslV | 1 |
| Cold stress | ||
| cspA | cold shock protein | 5 |
| Acid stress | ||
| atpA | F-type H+/Na+-transporting ATPase subunit alpha | 1 |
| atpB | F-type H+-transporting ATPase subunit a | 1 |
| atpE | F-type H+-transporting ATPase subunit c | 2 |
| atpF | F-type H+-transporting ATPase subunit b | 1 |
| atpH | F-type H+-transporting ATPase subunit delta | 1 |
| atpG | F-type H+-transporting ATPase subunit gamma | 1 |
| atpD | F-type H+/Na+-transporting ATPase subunit beta | 1 |
| atpC | F-type H+-transporting ATPase subunit epsilon | 1 |
| gadB | gadB, gadA, GAD; glutamate decarboxylase | 1 |
| nhaC | Na+:H+ antiporter, NhaC family | 1 |
| Bile tolerance | ||
| cbh | choloylglycine hydrolase | 1 |
| ppaC | manganese-dependent inorganic pyrophosphatase | 1 |
| cfa | cyclopropane-fatty-acyl-phospholipid synthase | 1 |
| Adhesion | ||
| mapA | maltose phosphorylase | 2 |
| lspA | lipoprotein signal peptidase II | 1 |
| tuf | elongation factor Tu | 1 |
| gpr | L-glyceraldehyde 3-phosphate reductase | 1 |
| tpiA | triosephosphate isomerase (TIM) | 1 |
| gapA | glyceraldehyde 3-phosphate dehydrogenase (phosphorylating) | 1 |
| bgaB | beta-galactosidase | 1 |
| srtA | sortase A | 1 |
| epsA | protein tyrosine kinase modulator | 2 |
| epsB | protein-tyrosine kinase | 2 |
| pgaC | poly-beta-1,6-N-acetyl-D-glucosamine synthase | 3 |
| eno | enolase | 2 |
| pgi | glucose-6-phosphate isomerase | 1 |
| LPXTG | surface protein (LPXTG motif) | 1 |
| epsH | Putative glycosyltransferase EpsH | 1 |
| Antioxidant | ||
| katE | catalase | 1 |
| fnr | ferredoxin/flavodoxin—NADP+ reductase | 5 |
| nrdH | glutaredoxin | 2 |
| gpx | glutathione peroxidase | 1 |
| gsr | glutathione reductase | 2 |
| mntH | manganese transport protein | 2 |
| mntA | manganese transport system ATP-binding protein | 1 |
| mntB | manganese transport system permease protein | 5 |
| mntC | manganese transport system substrate-binding protein | 2 |
| ndh | NADH dehydrogenase | 1 |
| npr | NADH peroxidase | 2 |
| poxL | pyruvate oxidase | 1 |
| tpx | thiol peroxidase | 1 |
| trxA | thioredoxin | 1 |
| trxB | thioredoxin reductase | 3 |
| nox | nadh oxidase | 4 |
| msrA | peptide-methionine (S)-S-oxide reductase | 2 |
| msrB | peptide-methionine (R)-S-oxide reductase | 1 |
| msrC | L-methionine (R)-S-oxide reductase | 1 |
| Immunomodulation | ||
| dltA | D-alanine—poly(phosphoribitol) ligase subunit 1 | 1 |
| dltB | membrane protein involved in D-alanine export | 1 |
| dltC | D-alanine—poly(phosphoribitol) ligase subunit 2 | 1 |
| dltD | D-alanine transfer protein | 1 |
| (a) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| CRISPR_ID | Start | End | Length (bp) | Orientation | Consensus Repeat | No. of CRISPRs with Same Repeat (Crisprdb) | Repeat Length | No. of Spacers | Evidence Level |
| Contig 2_1 | 480,884 | 481,711 | 827 | Forward | GTCTTGAATAGTAGTCATATCAAACAGGTTTAGAAC | 4 | 36 | 12 | 4 |
| Contig 5_1 | 73,501 | 73,616 | 115 | Forward | CTTGAACCAGCAAAGAGTTGTTGAACTGCACT | 0 | 32 | 1 | 1 |
| Contig 5_2 | 75,535 | 75,647 | 112 | Unknown | GAACCATCAGCCAACTGACTGACACCACT | 0 | 29 | 1 | 1 |
| (b) | |||||||||
| Sequence ID | Cas-Type/Subtype | Gene Status | System | Type | Begin | End | Strand | ||
| Contig 2_434 | cas2_TypeI-II-III | mandatory | CAS | CDS | 479,880 | 480,185 | + | ||
| Contig 2_433 | cas1_TypeII | forbidden | CAS-TypeIIU | CDS | 478,987 | 479,823 | + | ||
| Contig 2_432 | cas9_TypeII | forbidden | CAS-TypeIIU | CDS | 474,716 | 478,792 | + | ||
| Contig 2_435 | csn2_TypeIIA | forbidden | CAS-TypeIIA | CDS | 480,182 | 480,859 | + | ||
| Region | Region Length (kb) | Completeness | Score | Total Proteins | Region Position (bp) | Most Common Phage (Number of Matching Proteins) | GC% | attL/attR Sites | Integrase ORF Start–Stop |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 16.8 | intact | 100 | 31 | 377,986–394,877 | PHAGE_Entero_phiSHEF4_NC_042022(2) | 41.69 | + | 379,616–380,770 |
| 2 | 19.7 | Questionable | 70 | 22 | 262,426–282,208 | PHAGE_Entero_vB_EfaS_AL2_NC_042127(2) | 41.62 | + | 262,710–263,867 |
| 3 | 53.9 | intact | 150 | 56 | 155,716–209,682 | PHAGE_Lactob_Sha1_NC_019489(32) | 42.26 | + | 205,632–206,768 |
| Resistance | Description | KEGG_ID | Gene Name | Gene Location |
|---|---|---|---|---|
| Tetracycline resistance | Others | K18220 | tetM, tetO; ribosomal protection tetracycline resistance protein | contig00002_393 |
| Macrolide resistance | Transporters | K18231 | msr, vmlR; macrolide transport system ATP-binding/permease protein | contig00007_107 |
| K08217 | mef; MFS transporter, DHA3 family, macrolide efflux protein | contig00004_31 | ||
| Phenicol resistance | Acetyltransferases | K19271 | catA; chloramphenicol O-acetyltransferase type A [EC:2.3.1.28] | contig00001_343 |
| beta-Lactam resistance | Bla system [MD:M00627] | K17836 | penP; beta-lactamase class A [EC:3.5.2.6] | contig00004_310; contig00009_56 |
| Vancomycin resistance | D-Ala-D-Lac type [MD:M00651] | K07260 | vanY; zinc D-Ala-D-Ala carboxypeptidase [EC:3.4.17.14] | contig00003_219 |
| K08641 | vanX; zinc D-Ala-D-Ala dipeptidase [EC:3.4.13.22] | contig00008_133 | ||
| Cationic antimicrobial peptide (CAMP) resistance | dltABCD operon [MD:M00725] | K03367 | dltA; D-alanine―poly(phosphoribitol) ligase subunit 1 [EC:6.1.1.13] | contig00001_166 |
| K03739 | dltB; membrane protein involved in D-alanine export | contig00001_167 | ||
| K14188 | dltC; D-alanine―poly(phosphoribitol) ligase subunit 2 [EC:6.1.1.13] | contig00001_168; contig00006_124 | ||
| K03740 | dltD; D-alanine transfer protein | contig00001_169 | ||
| lysyl-phosphatidylglycerol (L-PG) synthase MprF [MD:M00726] | K14205 | mprF, fmtC; phosphatidylglycerol lysyltransferase [EC:2.3.2.3] | contig00008_63 | |
| Multidrug resistance | efflux pump AbcA [MD:M00700] | K18907 | norG; GntR family transcriptional regulator, regulator for abcA and norABC | contig00006_123 |
| K18104 | abcA, bmrA; ATP-binding cassette, subfamily B, bacterial AbcA/BmrA [EC:7.6.2.2] | contig00004_233; contig00005_131 | ||
| efflux pump NorB [MD:M00702] | K18907 | norG; GntR family transcriptional regulator, regulator for abcA and norABC | contig00006_123 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandasamy, S.; Yoo, J.; Yun, J.; Lee, K.-H.; Kang, H.-B.; Kim, J.-E.; Oh, M.-H.; Ham, J.-S. Probiogenomic In-Silico Analysis and Safety Assessment of Lactiplantibacillus plantarum DJF10 Strain Isolated from Korean Raw Milk. Int. J. Mol. Sci. 2022, 23, 14494. https://doi.org/10.3390/ijms232214494
Kandasamy S, Yoo J, Yun J, Lee K-H, Kang H-B, Kim J-E, Oh M-H, Ham J-S. Probiogenomic In-Silico Analysis and Safety Assessment of Lactiplantibacillus plantarum DJF10 Strain Isolated from Korean Raw Milk. International Journal of Molecular Sciences. 2022; 23(22):14494. https://doi.org/10.3390/ijms232214494
Chicago/Turabian StyleKandasamy, Sujatha, Jayeon Yoo, Jeonghee Yun, Kil-Ho Lee, Han-Byul Kang, Ji-Eun Kim, Mi-Hwa Oh, and Jun-Sang Ham. 2022. "Probiogenomic In-Silico Analysis and Safety Assessment of Lactiplantibacillus plantarum DJF10 Strain Isolated from Korean Raw Milk" International Journal of Molecular Sciences 23, no. 22: 14494. https://doi.org/10.3390/ijms232214494