
Comparative Analysis of Alternative Splicing in Two Contrasting Apple Cultivars Defense against Alternaria alternata Apple Pathotype Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
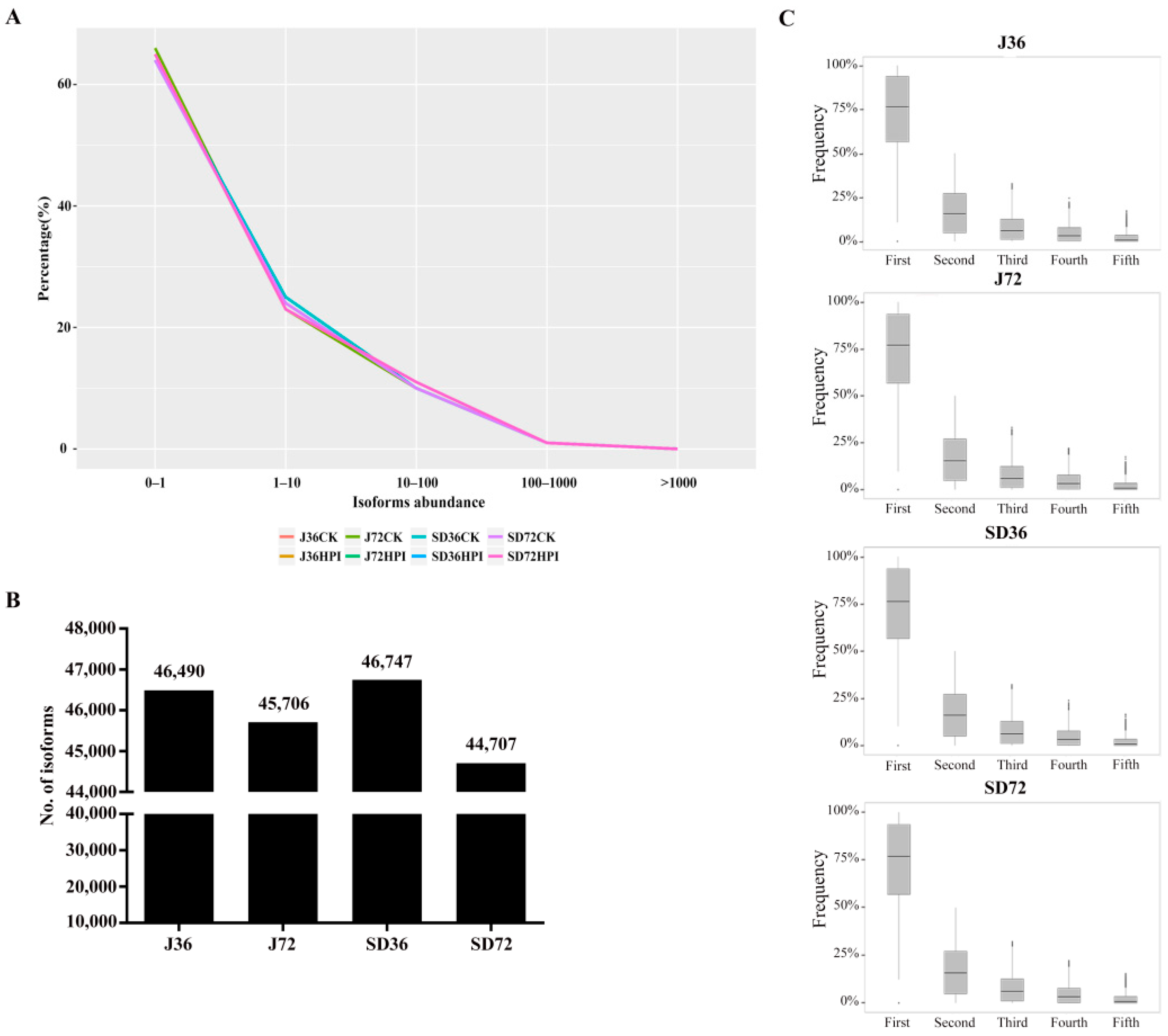
2.1. General Analysis of RNA-Seq Data
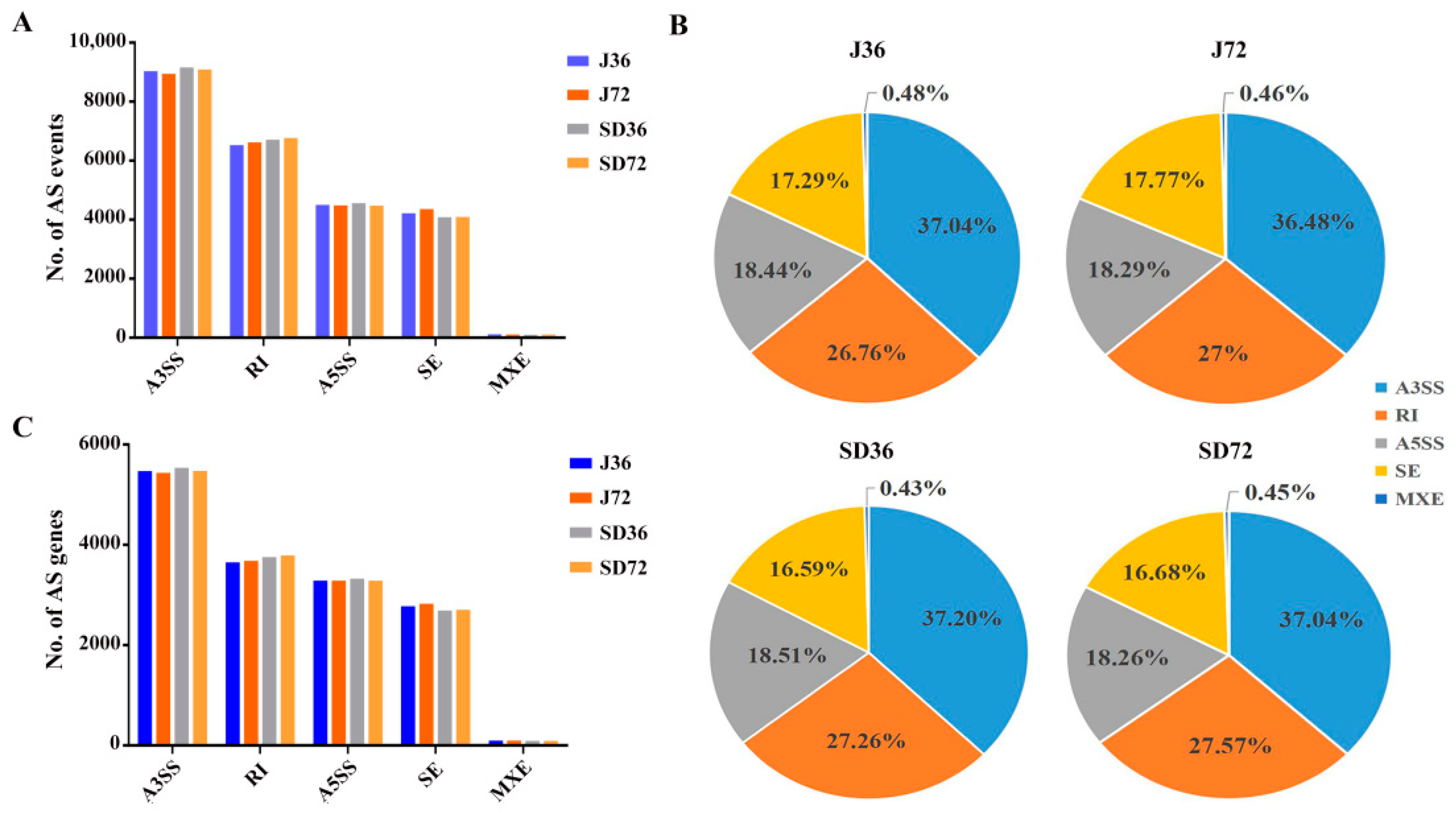
2.2. Detection and Classification of AS Events in Responding to A. alternata AP Infection in Apples
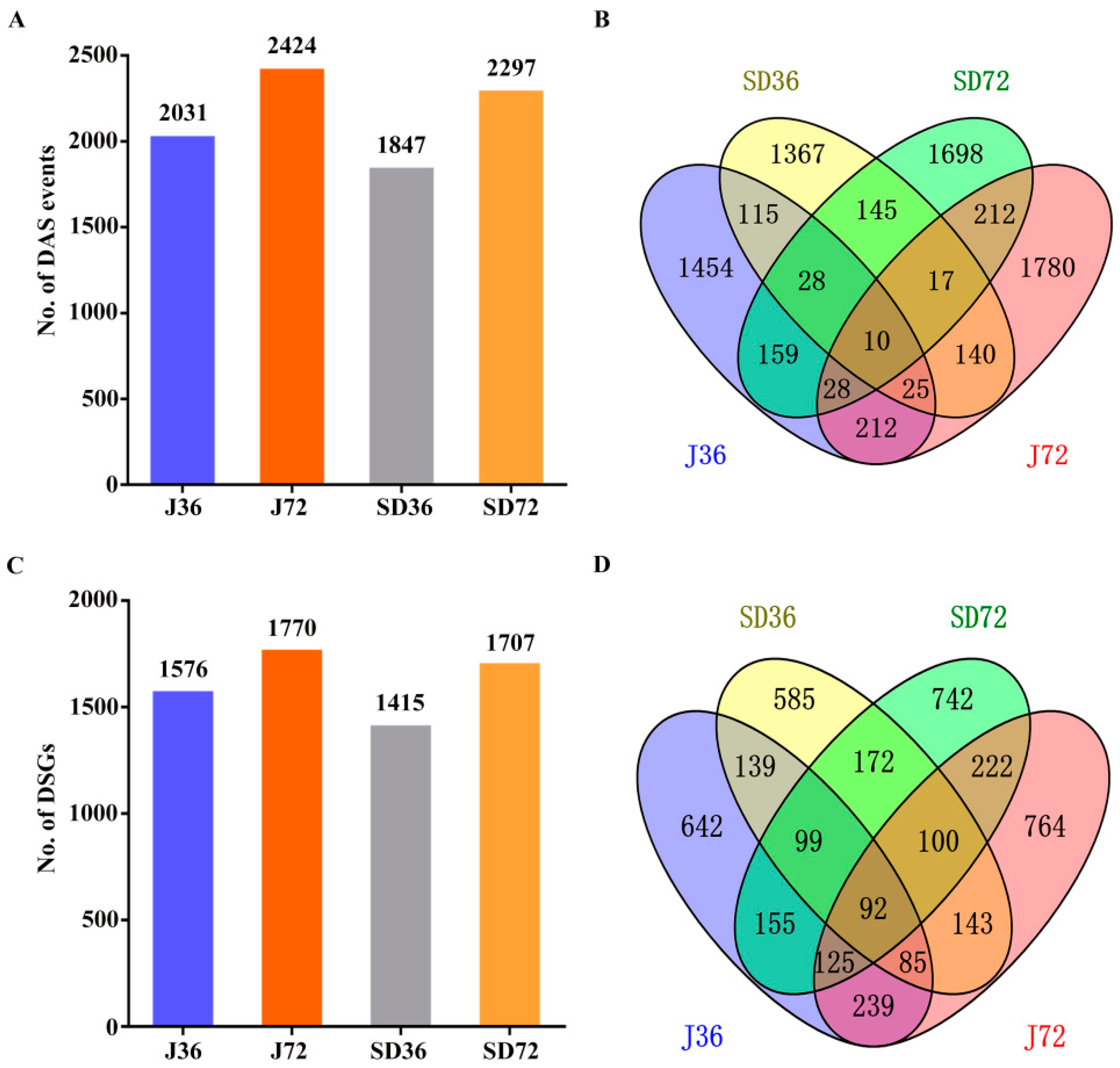
2.3. Identification and Analysis of DAS Events and Genes in Responding to A. alternata AP Infection
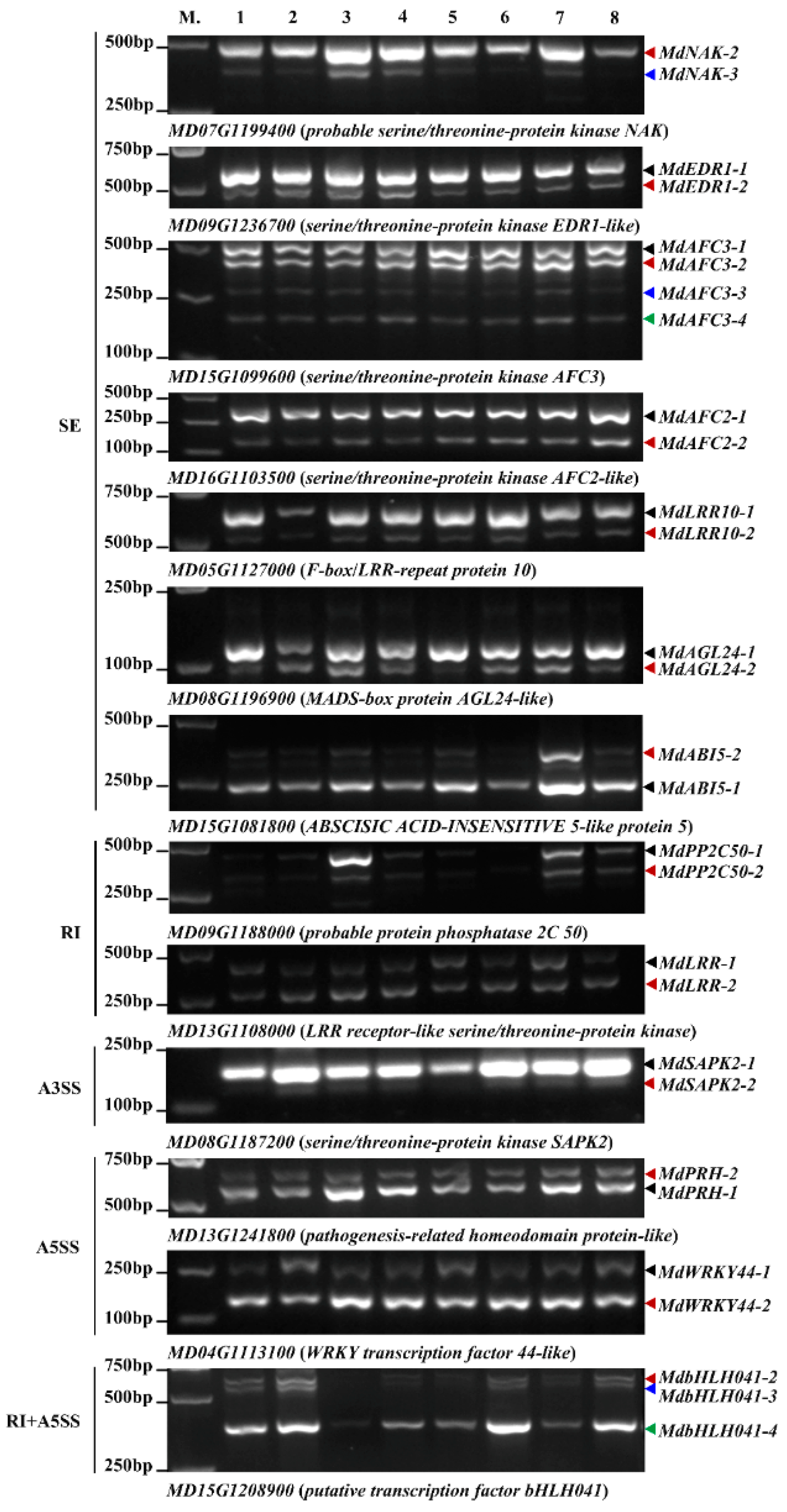
2.4. Validation of AS Events in Responding to A. alternata AP Infection
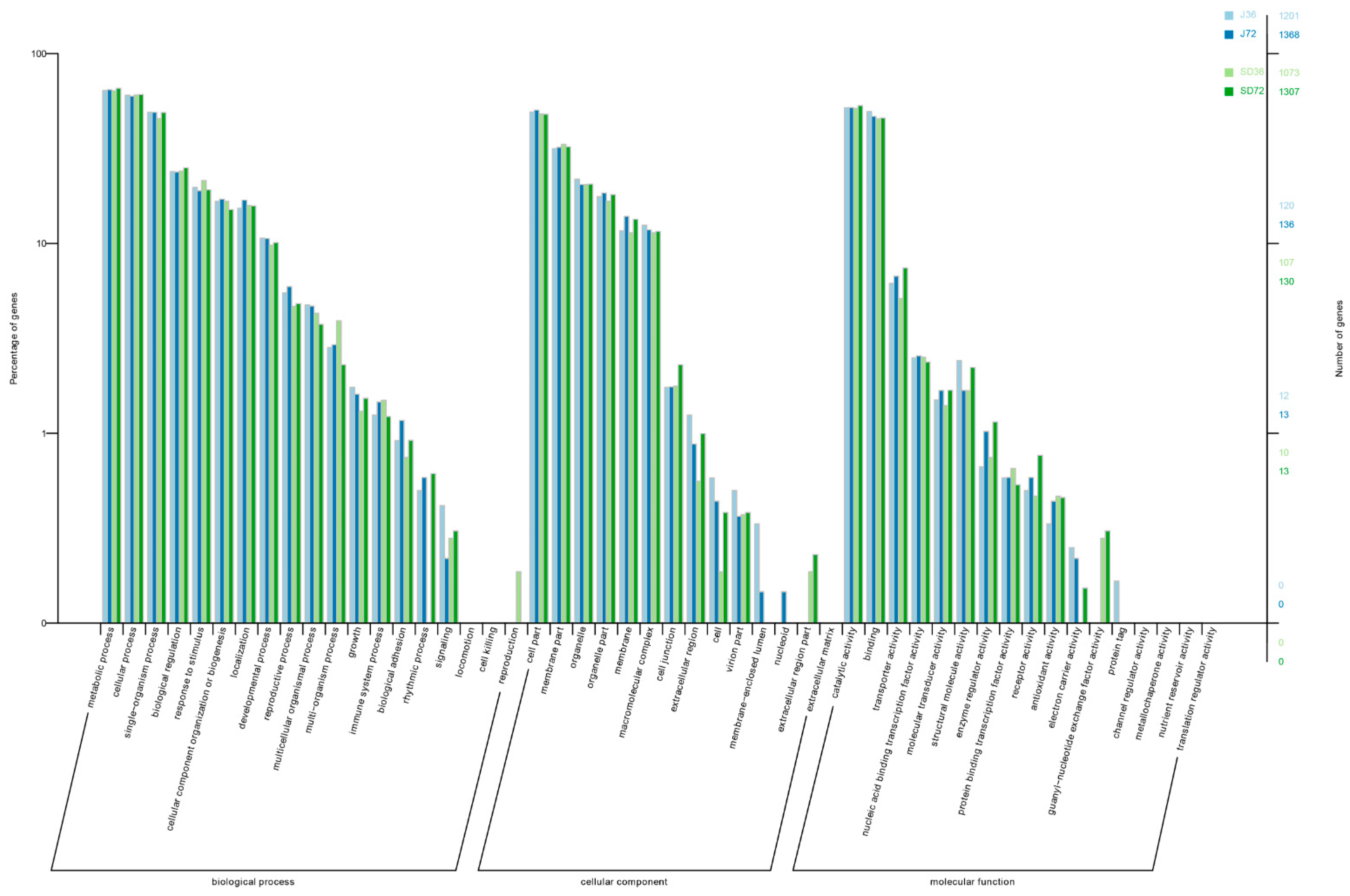
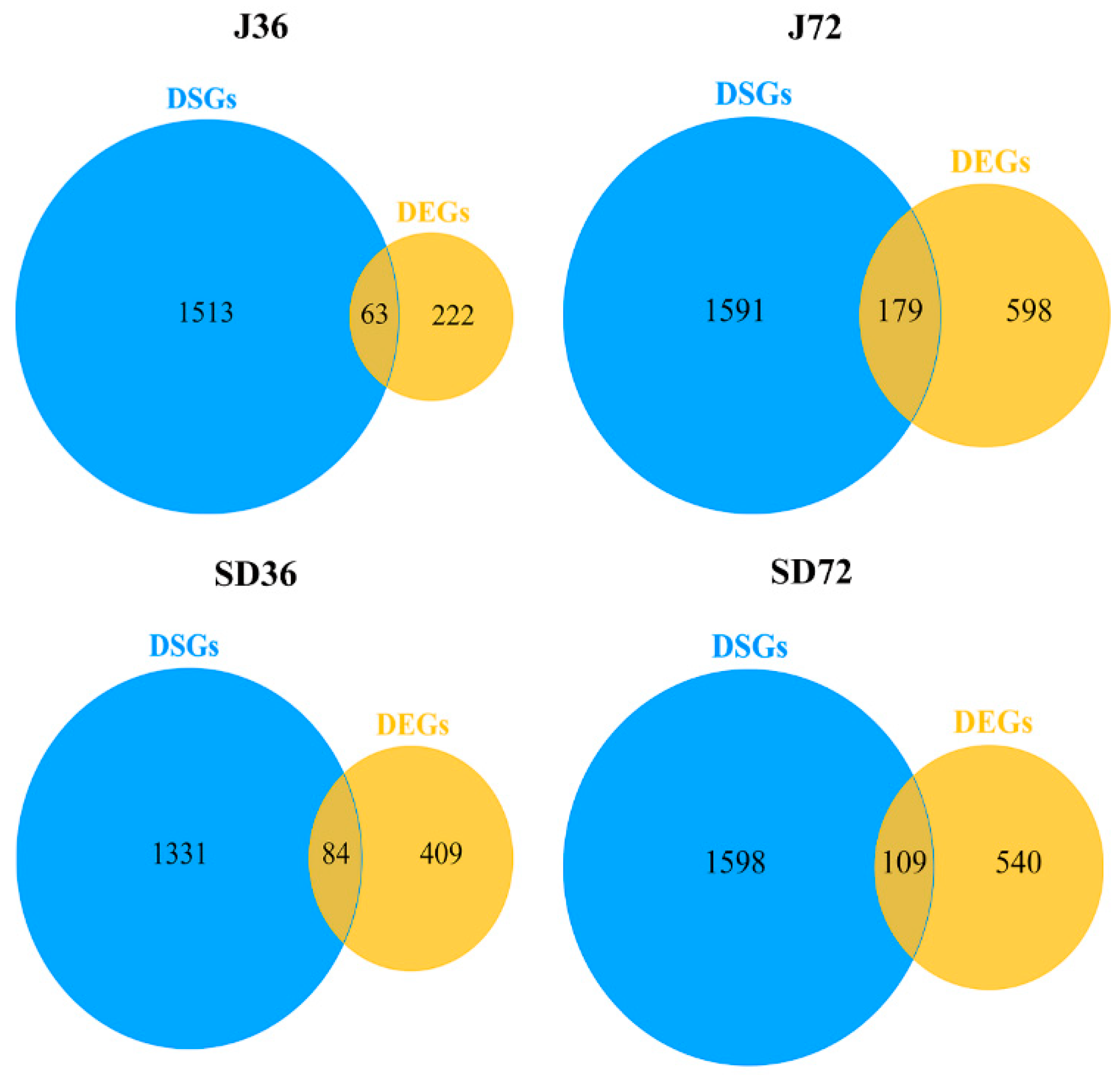
2.5. Comparative Analysis of the AS Genes in Responding to A. alternata AP Infection at AS and Transcriptional Levels
3. Discussion
3.1. AS Increased the Diversity of Transcripts in Apple
3.2. AS Is an Important Regulation Mechanism of Apple in Response to A. alternata AP Infection
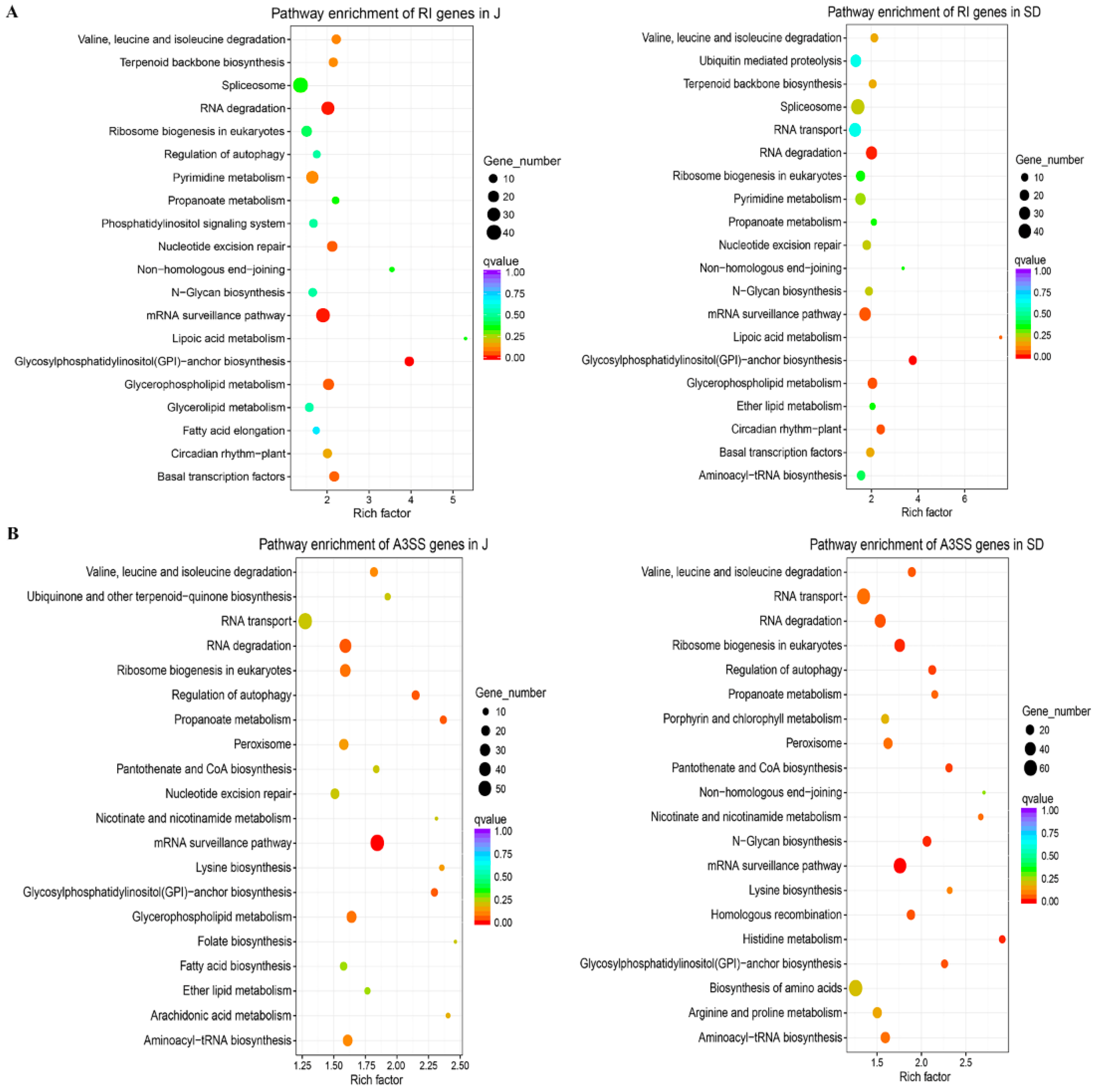
3.3. Considerable Number of Genes Occurring AS Changes Were Related to Stress Response
3.4. AS Provoked Specific Regulatory Functions during A. alternata AP Defensive Response
4. Materials and Methods
4.1. Plant Materials and Fungal Inoculation
4.2. RNA Extraction, RNA-Seq Libraries Construction and Sequencing
4.3. Detection and Classification of AS Events
4.4. Validation of AS Events
4.5. Analysis of Differentially Expressed Genes and Differentially Spliced Genes
4.6. Analysis of Gene Structure and Protein Domains
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AS | Alternative splicing |
| DAS | Differential alternative splicing |
| DSG | Differentially spliced gene |
| DEG | Differentially expressed gene |
| FPKM | Fragments per kilobase of transcript per million mapped reads |
| J | Jonathan |
| SD | Starking Delicious |
| HPI | Hours post inoculation |
| GO | Gene ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| SE | Skipped exon |
| RI | Retained intron |
| A3SS | Alternative 3′ splice sites |
| A5SS | Alternative 5′ splice sites |
| MXE | Mutually exclusive exon |
| A. alternata AP | Alternaria alternata apple pathotype |
| Bgt | Blumeria graminis f. sp. tritici |
| Pst | Puccinia striiformis f. sp. Tritici |
| M. oryzae | Magnaporthe oryzae |
| Xoo | Xanthomonas oryzae pv oryzae |
| SJs | Splicing junctions |
| CDS | Coding sequence |
| 5′-UTR | 5′ Un-translated region |
| 3′-UTR | 3′ Un-translated region |
| PTC | Premature stop codons |
| ORF | Open reading frame |
| NMD | Nonsense-mediated-decay |
| JA | Jasmonic acid |
| SA | Salicylic acid |
| ABA | Abscisic acid |
| PR | Pathogenesis-related |
| SR | Serine/arginine-rich |
| PDA | Potato dextrose agar |
References
- Zhuang, J.; Yao, Q.-H.; Xiong, A.-S.; Zhang, J. Isolation, phylogeny and expression patterns of AP2-like genes in apple (Malus × domestica Borkh). Plant Mol. Biol. Rep. 2011, 29, 209–216. [Google Scholar] [CrossRef]
- Li, Y.; Aldwinckle, H.S.; Sutton, T.; Tsuge, T.; Kang, G.D.; Cong, P.-H.; Cheng, Z.-M. Interactions of apple and the Alternariaalternata apple pathotype. Crit. Rev. Plant Sci. 2013, 32, 141–150. [Google Scholar] [CrossRef]
- Abe, K.; Iwanami, H.; Kotoda, N.; Moriya, S.; Takahashi, S. Evaluation of apple genotypes and Malus species for resistance to Alternaria blotch caused by Alternaria alternata apple pathotype using detached-leaf method. Plant Breed. 2010, 129, 208–218. [Google Scholar] [CrossRef]
- Johnson, R.D.; Johnson, L.; Itoh, Y.; Kodama, M.; Otani, H.; Kohmoto, K. Ciloning and characterization of a cyclic peptide synthetase gene from Alternaria alternata apple pathotype whose product is involved in AM-toxin synthesis and pathogenicity. Mol. Plant Microbe Interact. 2000, 13, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, D.; Otani, H.; Kodama, M. G protein signaling mediates developmental processes and pathogenesis of Alternaria alternata. Mol. Plant Microbe Interact. 2006, 19, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Harimoto, Y.; Tanaka, T.; Kodama, M.; Yamamoto, M.; Otani, H.; Tsuge, T. Multiple copies of AMT2 are prerequisite for the apple pathotype of Alternaria alternata to produce enough AM-toxin for expressing pathogenicity. J. Gen. Plant Pathol. 2008, 74, 222–229. [Google Scholar] [CrossRef]
- Zhang, C.-X.; Tian, Y.; Cong, P.-H. Proteome analysis of pathogen-responsive proteins from apple leaves induced by the Alternaria blotch Alternaria alternata. PLoS ONE 2015, 10, e0122233. [Google Scholar] [CrossRef]
- Zhu, L.M.; Ni, W.C.; Liu, S.; Cai, B.H.; Xing, H.; Wang, S.H. Transcriptomics analysis of apple leaves in response to Alternaria alternata apple pathotype infection. Front. Plant Sci. 2017, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Ni, W.C.; Zhu, L.M.; Sha, R.H.; Tao, J.M.; Cai, B.H.; Wang, S.H. Comparative iTRAQ proteomic profiling of susceptible and resistant apple cultivars infected by Alternaria alternata apple pathotype. Tree Genet. Genom. 2017, 13, 23. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, C.; Cong, P. Dynamics of growth regulators during infection of apple leaves by Alternaria alternata apple pathotype. Australasian Plant Pathol. 2012, 41, 247–253. [Google Scholar] [CrossRef]
- Reddy, A.S.N. Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 2007, 58, 267–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Lu, X.Y.; Wu, H.W.; Xie, Y.C.; Peng, Q.; Gu, L.F.; Wu, J.Y.; Wang, Y.C.; Reddy, A.S.N.; Dong, S.M. Phytophthora effectors modulate genome-wide alternative splicing of host mRNAs to reprogram plant immunity. Mol. Plant 2020, 13, 1470–1484. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-B.; Brendel, V. Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.F.; Liu, X.N.; Liu, C.H.; Liu, G.T.; Li, S.H.; Wang, L.J. Integrating omics and alternative splicing reveals insights into grape response to high temperature. Plant Physiol. 2017, 173, 1502–1518. [Google Scholar] [CrossRef] [Green Version]
- Braunschweig, U.; Gueroussov, S.; Plocik, A.M.; Graveley, B.R.; Blencowe, B.J. Dynamic integration of splicing within gene regulatory pathways. Cell 2013, 152, 1252–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Sandberg, R.; Luo, S.J.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Gao, Y.J.; Jia, H.T.; Liu, L.; Zhang, D.; Zhang, Z.X. Comparative transcriptomics uncovers alternative splicing changes and signatures of selection from maize improvement. BMC Genom. 2015, 16, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.S.N.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the alternative splicing landscape in plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef] [Green Version]
- Marquez, Y.; Brown, J.W.S.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.X.; Calixto, C.P.G.; Marquez, Y.; Venhuizen, P.; Tzioutziou, N.A.; Guo, W.B.; Spensley, M.; Entizne, J.C.; Lewandowska, D.; Have, S.T.; et al. A high quality Arabidopsis transcriptome for accurate transcript-level analysis of alternative splicing. Nucleic Acids Res. 2017, 45, 5061–5073. [Google Scholar] [CrossRef]
- Shen, Y.T.; Zhou, Z.K.; Wang, Z.; Li, W.Y.; Fang, C.; Wu, M.; Ma, Y.M.; Liu, T.F.; Kong, L.-A.; Peng, D.-L.; et al. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.L.; He, F.; Berkowitz, O.; Liu, J.X.; Cao, P.F.; Tang, M.; Shi, H.C.; Wang, W.J.; Li, Q.L.; Shen, Z.G.; et al. Alternative splicing plays a critical role in maintaining mineral nutrient homeostasis in rice (Oryza sativa). Plant Cell 2018, 30, 2267–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.Q.; Sun, M.; Wang, J.F.; Lei, M.; Li, C.J.; Zhao, D.J.; Huang, J.; Li, W.J.; Li, S.L.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019, 97, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.; Yu, F.; Gu, L.F.; Min, X.J. Expanding alternative splicing identification by integrating multiple sources of transcription data in tomato. Front. Plant Sci. 2019, 10, 689. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.S.; Qin, J.X.; Tian, X.J.; Xu, S.B.; Wang, Y.; Li, H.X.; Wang, X.M.; Peng, H.R.; Yao, Y.Y.; Hu, Z.R.; et al. Global profiling of alternative splicing landscape responsive to drought, heat and their combination in wheat (Triticum aestivum L.). Plant Biotechnol. J. 2017, 16, 714–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thatcher, S.R.; Zhou, W.G.; Leonard, A.; Wang, B.-B.; Beatty, M.; Zastrow-Hayes, G.; Zhao, X.Y.; Baumgarten, A.; Li, B.L. Genome-wide analysis of alternative splicing in Zea mays: Landscape and genetic regulation. Plant Cell 2014, 26, 3472–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-X.; Zhu, F.-Y.; Wang, F.-Z.; Ye, N.-H.; Gao, B.; Chen, X.; Zhao, S.-S.; Fan, T.; Cao, Y.-Y.; Liu, T.-Y.; et al. Alternative splicing and translation play important roles in hypoxic germination in rice. J. Exp. Bot. 2019, 70, 817–833. [Google Scholar] [CrossRef]
- Ishizawa, M.; Hashimoto, K.; Ohtani, M.; Sano, R.; Kurihara, Y.; Kusano, H.; Demura, T.; Matsui, M.; Sato-Nara, K. Inhibition of pre-mRNA splicing promotes root hair development in Arabidopsis thaliana. Plant Cell Physiol. 2019, 60, 1974–1985. [Google Scholar] [CrossRef]
- Li, L.; Hu, T.; Li, X.P.; Mu, S.H.; Cheng, Z.C.; Ge, W.; Gao, J. Genome-wide analysis of shoot growth-associated alternative splicing in moso bamboo. Mol. Genet. Genom. 2016, 291, 1695–1714. [Google Scholar] [CrossRef]
- Park, Y.-J.; Lee, J.-H.; Kim, J.Y.; Park, C.-M. Alternative RNA splicing expands the developmental plasticity of flowering transition. Front. Plant Sci. 2019, 10, 606. [Google Scholar] [CrossRef]
- Yuan, H.Z.; Yu, H.M.; Huang, T.; Shen, X.J.; Xia, J.; Pang, F.H.; Wang, J.; Zhao, M.Z. The complexity of the Fragaria x ananassa (octoploid) transcriptome by single-molecule long-read sequencing. Hortic. Res. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Y.; Mi, X.Z.; Zhao, S.Q.; Zhu, J.Y.; Guo, R.; Xia, X.B.; Liu, L.; Liu, S.R.; Wei, C.L. Comprehensive profiling of alternative splicing landscape during cold acclimation in tea plant. BMC Genom. 2020, 21, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calixto, C.P.G.; Guo, W.B.; James, A.B.; Tzioutziou, N.A.; Entizne, J.C.; Panter, P.E.; Knight, H.; Nimmo, H.G.; Zhang, R.X.; Brown, J.W.S. Rapid and dynamic alternative splicing impacts the Arabidopsis cold response transcriptome. Plant Cell 2018, 30, 1424–1444. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Liang, J.H.; Wang, C.P.; Ding, L.P.; Zhao, X.; Cao, X.; Xu, S.J.; Teng, N.J.; Yi, M.F. Alternative splicing provides a mechanism to regulate LlHSFA3 function in response to heat stress in lily. Plant Physiol. 2019, 181, 1651–1667. [Google Scholar] [CrossRef]
- Zhu, G.Z.; Li, W.X.; Zhang, F.; Guo, W.Z. RNA-seq analysis reveals alternative splicing under salt stress in cotton, Gossypium davidsonii. BMC Genom. 2018, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Cui, P.; Wang, Z.Y.; Zhang, S.D.; Ali, S.; Xiong, L.M. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genom. 2014, 15, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.F.; Xiao, B.Z. Comparative alternative splicing analysis of two contrasting rice cultivars under drought stress and association of differential splicing genes with drought response QTLs. Euphytica 2018, 214, 73. [Google Scholar] [CrossRef]
- Lee, J.S.; Gao, L.X.; Guzman, L.M.; Rieseberg, L.H. Genome-wide expression and alternative splicing in domesticated sunflowers (Helianthus annuus L.) under flooding stress. Agronomy 2021, 11, 92. [Google Scholar] [CrossRef]
- Wu, F.M.; Deng, L.; Zhai, Q.Z.; Zhao, J.H.; Chen, Q.; Li, C.Y. Mediator subunit MED25 couples alternative splicing of JAZ genes with fine-tuning of jasmonate signaling. Plant Cell 2020, 32, 429–448. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.-Y.; Chen, M.X.; Ye, N.-H.; Shi, L.; Ma, K.-L.; Yang, J.-F.; Cao, Y.-Y.; Zhang, Y.J.; Yoshida, T.; Fernie, A.R.; et al. Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in Arabidopsis seedlings. Plant J. 2017, 91, 518–533. [Google Scholar] [CrossRef]
- Zhang, J.K.; Jiao, P.; Zhang, C.; Tong, X.L.; Wei, Q.P.; Xu, L.F. Apple NPR1 homologs and their alternative splicing forms may contribute to SA and disease responses. Tree Genet. Genom. 2016, 12, 92. [Google Scholar] [CrossRef]
- Wang, Y.C.; Xu, J.Y.; Ge, M.; Ning, L.H.; Hu, M.M.; Zhao, H. High-resolution profile of transcriptomes reveals a role of alternative splicing for modulating response to nitrogen in maize. BMC Genom. 2020, 21, 353. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Kakei, Y.; Shimada, Y.; Fujiwara, T. Genome-wide analysis of specific alterations in transcript structure and accumulation caused by nutrient deficiencies in Arabidopsis thaliana. Plant J. 2017, 91, 741–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.X.; Chen, M.X.; Zhu, F.Y.; Fan, T.; Zhang, J.H.; Lo, C. Alternative splicing is a Sorghum bicolor defense response to fungal infection. Planta 2020, 251, 14. [Google Scholar] [CrossRef] [PubMed]
- Sampangi-Ramaiah, M.H.; Ravishankar, K.V.; Nataraja, K.N.; Shaanker, R.U. Endophytic fungus, Fusarium sp. reduces alternative splicing events in rice plants under salinity stress. Plant Physiol. Rep. 2019, 24, 487–495. [Google Scholar] [CrossRef]
- Palma, M.D.; Salzano, M.; Villano, C.; Aversano, R.; Lorito, M.; Ruocco, M.; Docimo, T.; Piccinelli, A.L.; D’Agostino, N.; Tucci, M. Transcriptome reprogramming, epigenetic modifications and alternative splicing orchestrate the tomato root response to the beneficial fungus Trichoderma harzianum. Hortic. Res. 2019, 6, 5. [Google Scholar] [CrossRef]
- Zorin, E.A.; Afonin, A.M.; Kulaeva, O.A.; Gribchenko, E.S.; Shtark, O.Y.; Zhukov, V.A. Transcriptome analysis of alternative splicing events induced by arbuscular mycorrhizal fungi (Rhizophagus irregularis) in pea (Pisum sativum L.) roots. Plants 2020, 9, 1700. [Google Scholar] [CrossRef]
- Ma, J.-Q.; Wei, L.-J.; Lin, A.; Zhang, C.; Sun, W.; Yang, B.; Lu, K.; Li, J.-N. The alternative splicing landscape of Brassica napus infected with Leptosphaeria maculans. Genes 2019, 10, 296. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Mao, R.; Wang, Y.Z.; Zhang, L.; Wang, C.Y.; Lv, S.K.; Liu, X.L.; Wang, Y.J.; Ji, W.Q. Transcriptome-wide alternative splicing modulation during plant-pathogen interactions in wheat. Plant Sci. 2019, 288, 110160. [Google Scholar] [CrossRef]
- Liu, J.Q.; Chen, X.J.; Liang, X.X.; Zhou, X.G.; Yang, F.; Liu, J.; He, S.Y.; Guo, Z.J. Alternative splicing of rice WRKY62 and WRKY76 transcription factor genes in pathogen defense. Plant Physiol. 2016, 171, 1427–1442. [Google Scholar] [CrossRef]
- Ma, H.R.; Wang, F.; Wang, W.J.; Yin, G.Y.; Zhang, D.Y.; Ding, Y.Q.; Timko, M.P.; Zhang, H.B. Alternative splicing of basic chitinase gene PR3b in the low-nicotine mutants of Nicotiana tabacum L. cv. Burley 21. J. Exp. Bot. 2016, 67, 5799–5809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo, S.; Peris-Peris, C.; Siré, C.; Moreno, A.B.; Donaire, L.; Zytnicki, M.; Notredame, C.; Llave, C.; Segundo, B.S. Identification of a novel microRNA (miRNA) from rice that targets an alternatively spliced transcript of the Nramp6 (Natural resistance-associated macrophage protein 6) gene involved in pathogen resistance. New Phytol. 2013, 199, 212–227. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.L.; Sivamani, E.; Azhakanandam, K.; Samadder, P.; Li, X.G.; Qu, R.D. Gene expression enhancement mediated by the 5′UTR intron of the rice rubi3 gene varied remarkably among tissues in transgenic rice plants. Mol. Genet. Genom. 2008, 279, 563–572. [Google Scholar] [CrossRef]
- Chung, B.Y.; Simons, C.; Firth, A.E.; Brown, C.M.; Hellens, R.P. Effect of 5′UTR introns on gene expression in Arabidopsis thaliana. BMC Genom. 2006, 7, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.Q.; Wang, Y.; Qiu, C.; Qian, W.J.; Xie, H.; Ding, Z.T. Alternative splicing in tea plants was extensively triggered by drought, heat and their combined stresses. PeerJ 2020, 8, e8258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, J.L.; Tong, L.; Fang, Z.Y.; Yang, L.M.; Zhuang, M.; Zhang, Y.Y.; Lv, H.H. Global survey of the full-length cabbage transcriptome (Brassica oleracea var. capitata L.) reveals key alternative splicing events involved in growth and disease response. Int. J. Mol. Sci. 2021, 22, 10443. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Wang, Y.; Fang, Z.Y.; Zhuang, M.; Zhang, Y.Y.; Lv, H.H.; Liu, Y.M.; Li, Z.S.; Yang, L.M. Comparative transcriptome analysis of cabbage (Brassica oleracea var. capitata) infected by Plasmodiophora brassicae reveals drastic defense response at secondary infection stage. Plant Soil 2019, 443, 167–183. [Google Scholar] [CrossRef]
- Sun, Y.; Hou, H.; Song, H.T.; Lin, K.; Zhang, Z.H.; Hu, J.L.; Pang, E. The comparison of alternative splicing among the multiple tissues in cucumber. BMC Plant Biol. 2018, 18, 5. [Google Scholar] [CrossRef] [Green Version]
- Howard, B.E.; Hu, Q.W.; Babaoglu, A.C.; Chandra, M.; Borghi, M.; Tan, X.P.; He, L.Y.; Winter-Sederoff, H.; Gassmann, W.; Veronese, P.; et al. High-throughput RNA sequencing of Pseudomonas-infected Arabidopsis reveals hidden transcriptome complexity and novel splice variants. PLoS ONE 2013, 8, e74183. [Google Scholar] [CrossRef]
- Qin, N.; Zhang, R.Z.; Zhang, M.C.; Niu, Y.; Fu, S.Y.; Wang, Y.S.; Wang, D.D.; Chen, Y.; Zhao, C.Z.; Chen, Q.; et al. Global profiling of dynamic alternative splicing modulation in Arabidopsis root upon Ralstonia solanacearum infection. Genes 2020, 11, 1078. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-Q.; Xu, W.; Xu, F.; Lin, A.; Sun, W.; Jiang, H.-H.; Lu, K.; Li, J.-N.; Wei, L.-J. Differential alternative splicing genes and isoform regulation networks of rapeseed (Brassica napus L.) infected with Sclerotinia sclerotiorum. Genes 2020, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Ganie, S.A.; Reddy, A.S.N. Stress-induced changes in alternative splicing landscape in rice:functional significance of splice isoforms in stress tolerance. Biology 2021, 10, 309. [Google Scholar] [CrossRef] [PubMed]
- Laloum, T.; Martín, G.; Duque, P. Alternative splicing control of abiotic stress responses. Trends Plant Sci. 2018, 23, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.M.; Tang, F.; Zhu, H.Y. Alternative splicing in plant immunity. Int. J. Mol. Sci. 2014, 15, 10424–10445. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.Z.; Christiansen, K.M.; Innes, R.W. Regulation of plant disease resistance, stress responses, cell death, and ethylene signaling in Arabidopsis by the EDR1 protein kinase. Plant Physiol. 2005, 138, 1018–1026. [Google Scholar] [CrossRef] [Green Version]
- Serrano, I.; Gu, Y.N.; Qi, D.; Dubiella, U.; Innes, R.W. The Arabidopsis EDR1 protein kinase negatively regulates the ATL1 E3 ubiquitin ligase to suppress cell death. Plant Cell 2014, 26, 4532–4546. [Google Scholar] [CrossRef] [Green Version]
- Wawrzynska, A.; Rodibaugh, N.L.; Innes, R.W. Synergistic activation of defense responses in Arabidopsis by simultaneous loss of the GSL5 callose synthase and the EDR1 protein kinase. Mol. Plant Microbe Interact. 2010, 23, 578–584. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, K.M.; Gu, Y.N.; Rodibaugh, N.; Innes, R.W. Negative regulation of defence signalling pathways by the EDR1 protein kinase. Mol. Plant Pathol. 2011, 12, 746–758. [Google Scholar] [CrossRef]
- Lin, J.Y.; Shi, J.J.; Zhang, Z.H.; Zhong, B.J.; Zhu, Z.Q. Plant AFC2 kinase desensitizes thermomorphogenesis through modulation of alternative splicing. iScience 2022, 25, 104051. [Google Scholar] [CrossRef]
- Golovkin, M.; Reddy, A.S.N. An SC35-like protein and a novel serine/arginine-rich protein interact with Arabidopsis U1-70K protein. J. Biol. Chem. 1999, 274, 36428–36438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, M.; Mahuku, G.; Swennen, R. Comparative transcriptome and expression profiling of resistant and susceptible banana cultivars during infection by Fusarium oxysporum. Int. J. Mol. Sci. 2021, 22, 3002. [Google Scholar] [CrossRef] [PubMed]
- Meraj, T.A.; Fu, J.Y.; Raza, M.A.; Zhu, C.Y.; Shen, Q.Q.; Xu, D.B.; Wang, Q. Transcriptional factors regulate plant stress responses through mediating secondary metabolism. Genes 2020, 11, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, S.H.; Anand, S.; Singh, B.; Bohra, A.; Josh, R. WRKY transcription factors and plant defense responses:latest discoveries and future prospects. Plant Cell Rep. 2021, 40, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Li, X.S.; Wen, X.J.; Zhang, Y.; Ding, Y.; Zhang, Y.H.; Gao, B.; Zhang, D.Y. PacBio full-length transcriptome of wild apple (Malus sieversii) provides insights into canker disease dynamic response. BMC Genom. 2021, 22, 52. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-H.; Kim, D.-H.; Jung, J.-A.; Lee, J.-Y. Alternative splicing of the basic helix-loop-helix transcription factor gene CmbHLH2 affects anthocyanin biosynthesis in ray florets of chrysanthemum (Chrysanthemum morifolium). Front. Plant Sci. 2021, 12, 669315. [Google Scholar] [CrossRef]
- Guo, J.L.; Ling, H.; Ma, J.J.; Chen, Y.; Su, Y.C.; Lin, Q.L.; Gao, S.W.; Wang, H.B.; Que, Y.X.; Xu, L.P. A sugarcane R2R3-MYB transcription factor gene is alternatively spliced during drought stress. Sci. Rep. 2017, 7, 41922. [Google Scholar] [CrossRef] [Green Version]
- Seok, H.-Y.; Ha, J.M.; Lee, S.-Y.; Bae, H.; Moon, Y.-H. Two alternative splicing variants of AtERF73/HRE1, HRE1α and HRE1β, have differential transactivation activities in Arabidopsis. Int. J. Mol. Sci. 2020, 21, 6984. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.F.; Zhang, H.; Wang, L.C.; Zhu, Z.G.; Gao, J.P.; Li, C.S.; Zhu, Y. High temperature inhibits the accumulation of storage materials by inducing alternative splicing of OsbZIP58 during filling stage in rice. Plant Cell Environ. 2020, 43, 1879–1896. [Google Scholar] [CrossRef]
- Zou, M.J.; Guan, Y.C.; Ren, H.B.; Zhang, F.; Chen, F. Characterization of alternative splicing products of bZIP transcription factors OsABI5. Biochem. Biophys. Res. Commun. 2007, 360, 307–313. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 13 November 2022).
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- ALove, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrian, A.; Rahnenfuhrer, J. topGO; Bioconductor: Buffalo, NY, USA, 2017. [Google Scholar]
- Hu, B.; Jin, J.P.; Guo, A.-Y.; Zhang, H.; Luo, J.C.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; He, Y.; Zeng, X.; Cai, B.; Qu, S.; Wang, S. Comparative Analysis of Alternative Splicing in Two Contrasting Apple Cultivars Defense against Alternaria alternata Apple Pathotype Infection. Int. J. Mol. Sci. 2022, 23, 14202. https://doi.org/10.3390/ijms232214202
Zhou T, He Y, Zeng X, Cai B, Qu S, Wang S. Comparative Analysis of Alternative Splicing in Two Contrasting Apple Cultivars Defense against Alternaria alternata Apple Pathotype Infection. International Journal of Molecular Sciences. 2022; 23(22):14202. https://doi.org/10.3390/ijms232214202
Chicago/Turabian StyleZhou, Tingting, Youlei He, Xianqi Zeng, Binhua Cai, Shenchun Qu, and Sanhong Wang. 2022. "Comparative Analysis of Alternative Splicing in Two Contrasting Apple Cultivars Defense against Alternaria alternata Apple Pathotype Infection" International Journal of Molecular Sciences 23, no. 22: 14202. https://doi.org/10.3390/ijms232214202