

The PPARα Regulation of the Gut Physiology in Regard to Interaction with Microbiota, Intestinal Immunity, Metabolism, and Permeability


Abstract
:1. Introduction
2. The Role of PPARα in the Gastrointestinal Mucosa: Modulation of Metabolism and Immunity
2.1. The Role of PPARa in Immunotolerance of Dietary Antigens
2.2. The Role of PPARα in Pathophysiology of Colitis
2.3. The Role of PPARα in the Production of Antimicrobial Peptides and Paneth Cell Functions
3. PPARα-Mediated Modulation of Gut Microbiota Composition
3.1. The PPARa Influence on the Microbiota Profile
3.2. The PPARa Agonist as Modulators of Gut Microbiota Diversity

{kind=link}
{kind=link}
| Model/Agent | Outcome | Reference |
|---|---|---|
| Ppara −/− mice | Increased population of segmented filamentous bacteria (SFB), TM7 bacteria | [18] |
| Oleoylethanolamide (OEA), an endogenous PPARα agonist, mice | A trend of Bacillus, Lactobacillus decrease, Bacteroides, Prevotella, Parabacteroides increase, decreased Firmicutes/Bacteroidetes ratio | [42] |
| Wy-16434, a synthetic PPARα agonist, mice on high-fructose diet | Reverse of dysbiosis induced by a high-fructose diet, decreased Proteobacteria, increased Actinobacteria counts | [53,54] |
4. Modulation of PPAR Activity by the Various Taxa of Commensal Bacteria
| Species/Strains | Active Form or Metabolite | Molecular Target | Reference |
|---|---|---|---|
| Lactobacillus plantarum (Lactiplantibacillus plantarum) AKU1009a | α-linoleic acid metabolites, keto- and hydroxy-octadecenoic acid species | PPARα, PPARγ | [61,62] |
| Lactobacillus kefiri (Lentilactobacillus kefiri) DH5 | Live bacteria | PPARα | [66] |
| Lactobacillus amylovorus CP1563 | Mill-fragmented bacteria | PPARα | [67,68] |
| Lactobacillus plantarum (Lactiplantibacillus plantarum) FRT10 | Live bacteria | PPARα | [69] |
| Bacillus subtilis + Enterococcus faecium | Live bacteria | PPARα | [70] |
| Bacteroides acidifaciens JCM10556 | Live bacteria | PPARα | [71] |
| Faecalibacterium prausnitzii | Extracellular vesicles (EVs) | PPARα, β, γ, tight junction proteins | [72] |
| propionate | PPARα | [75] | |
| Akkremansia muciniphila | Live bacteria, autoclaved bacteria | PPARα/RXRα | [82,84] |
| Akkermansia muciniphila | Live bacteria, 1-PG, 2-PG | PPARα | [94] |
| Akkermansia muciniphila | Pasteurized bacteria, Amuc_1100 | TLR2 | [95] |
| Lactobacillus plantarum (Lactiplantibacillus plantarum) NCIMB8826 | Live bacteria | PPARα | [96] |
5. PPARα and the Gut Microbiota Composition—Implications for Total Energy Expenditure
6. The PPARα-Mediated Alterations in Metabolism and Their Implications to Host-Microbiota Interactions
7. Interplay between PPARα, Nitric Oxide Production and Gut Microbiota
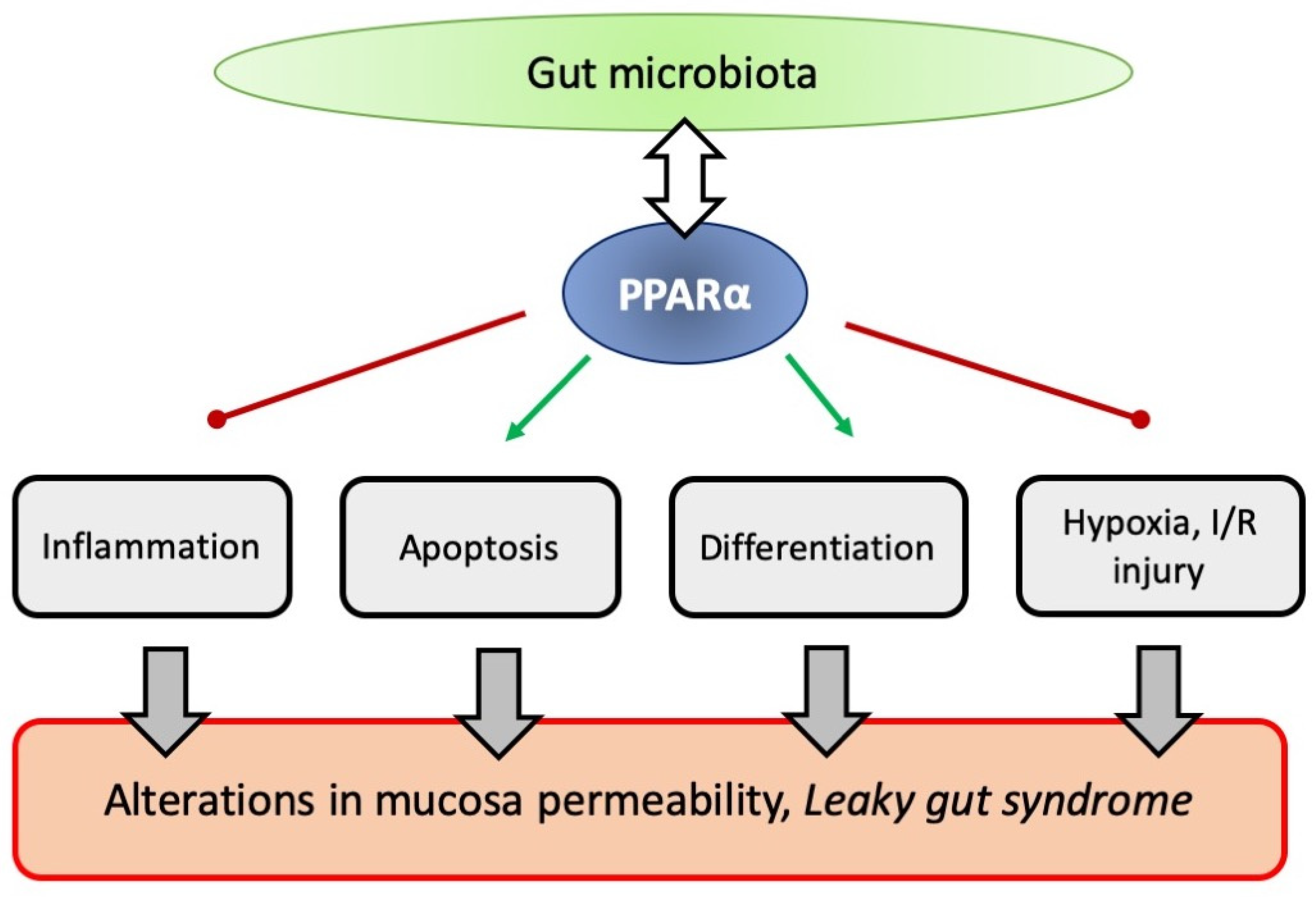
8. PPARα and Intestinal Permeability
8.1. PPARα Knock-out Mice and Intestinal Permeability
8.2. PPARα Action on Intestinal Barrier Function and Inflammation
8.3. PPARα Action on Intestinal Regeneration and Its Possible Influence on Gut Permeability
8.4. PPARα Action on Intestinal Permeability and Hypoxia
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Grabacka, M.; Pierzchalska, M.; Płonka, P.M.; Pierzchalski, P. The Role of PPAR Alpha in the Modulation of Innate Immunity. Int. J. Mol. Sci. 2021, 22, 10545. [Google Scholar] [CrossRef] [PubMed]
- Bünger, M.; Bosch, H.M.V.D.; Van Der Meijde, J.; Kersten, S.; Hooiveld, G.J.E.J.; Muller, M. Genome-wide analysis of PPARα activation in murine small intestine. Physiol. Genom. 2007, 30, 192–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARα Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e4. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.M.; Cho, D.H.; Youakim, A.; Bradley, M.B.; Lee, J.S.; Framson, P.E.; Nepom, G. Highly polarized HLA class II antigen processing and presentation by human intestinal epithelial cells. J. Clin. Investig. 1998, 102, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, V.; Goddeeris, B.; Cox, E. The role of enterocytes in the intestinal barrier function and antigen uptake. Microbes Infect. 2005, 7, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Strobel, S.; Mowat, A.M. Immune responses to dietary antigens: Oral tolerance. Immunol. Today 1998, 19, 173–181. [Google Scholar] [CrossRef]
- Mueller, D.; Jenkins, M.; Schwartz, R.H. Clonal Expansion Versus Functional Clonal Inactivation: A Costimulatory Signalling Pathway Determines the Outcome of T Cell Antigen Receptor Occupancy. Annu. Rev. Immunol. 1989, 7, 445–480. [Google Scholar] [CrossRef]
- Chen, Y.; Inobe, J.; Weiner, H.L. Induction of oral tolerance to myelin basic protein in CD8-depleted mice: Both CD4+ and CD8+ cells mediate active suppression. J. Immunol. 1995, 155, 910–916. [Google Scholar]
- Lelouard, H.; Fallet, M.; de Bovis, B.; Méresse, S.; Gorvel, J. Peyer’s Patch Dendritic Cells Sample Antigens by Extending Dendrites Through M Cell-Specific Transcellular Pores. Gastroenterology 2012, 142, 592–601.e3. [Google Scholar] [CrossRef]
- Jakobsen, M.A.; Petersen, R.K.; Kristiansen, K.; De Lange, M.; Lillevang, S.T. Peroxisome Proliferator-Activated Receptor alpha, delta, gamma1 and gamma2 Expressions are Present in Human Monocyte-Derived Dendritic Cells and Modulate Dendritic Cell Maturation by Addition of Subtype-Specific Ligands. Scand. J. Immunol. 2006, 63, 330–337. [Google Scholar] [CrossRef]
- Shi, H.-Y.; Ge, J.-B.; Fang, W.-Y.; Yao, K.; Sun, A.-J.; Huang, R.-C.; Jia, Q.-Z.; Wang, K.-Q.; Zou, Y.-Z.; Cao, X.-T. Peroxisome proliferator-activated receptor α agonist attenuates oxidized-low density lipoprotein induced immune maturation of human monocyte-derived dendritic cells. Chin. Med. J. 2008, 121, 1747–1750. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, S.; Reiser, G. Role of the peroxisome proliferator-activated receptors (PPAR)-α, β/δ and γ triad in regulation of reactive oxygen species signaling in brain. Biol. Chem. 2013, 394, 1553–1570. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.-T.; Nishiyama, K.; Matsuo, Y.; Kuwamura, M.; Morioka, A.; Nakajima, H.; Takeuchi, T. PPARα contributes to colonic protection in mice with DSS-induced colitis. Int. Immunopharmacol. 2010, 10, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Basso, P.J.; Sales-Campos, H.; Nardini, V.; Duarte-Silva, M.; Alves, V.B.F.; Bonfá, G.; Rodrigues, C.C.; Ghirotto, B.; Chica, J.E.L.; Nomizo, A.; et al. Peroxisome Proliferator-Activated Receptor Alpha Mediates the Beneficial Effects of Atorvastatin in Experimental Colitis. Front. Immunol. 2021, 12, 618365. [Google Scholar] [CrossRef] [PubMed]
- Katkar, G.D.; Sayed, I.M.; Anandachar, M.S.; Castillo, V.; Vidales, E.; Toobian, D.; Usmani, F.; Sawires, J.R.; Leriche, G.; Yang, J.; et al. Artificial intelligence-rationalized balanced PPARα/γ dual agonism resets dysregulated macrophage processes in inflammatory bowel disease. Commun. Biol. 2022, 5, 231. [Google Scholar] [CrossRef]
- Lee, J.W.; Bajwa, P.J.; Carson, M.J.; Jeske, D.R.; Cong, Y.; Elson, C.O.; Lytle, C.; Straus, D.S. Fenofibrate Represses Interleukin-17 and Interferon-γ Expression and Improves Colitis in Interleukin-10–Deficient Mice. Gastroenterology 2007, 133, 108–123. [Google Scholar] [CrossRef]
- Otagiri, S.; Ohnishi, S.; Ohara, M.; Fu, Q.; Yamamoto, K.; Yamamoto, K.; Katsurada, T.; Sakamoto, N. Oleoylethanolamide Ameliorates Dextran Sulfate Sodium-Induced Colitis in Rats. Front. Pharmacol. 2020, 11, 1277. [Google Scholar] [CrossRef]
- Manoharan, I.; Suryawanshi, A.; Hong, Y.; Ranganathan, P.; Shanmugam, A.; Ahmad, S.; Swafford, D.; Manicassamy, B.; Ramesh, G.; Koni, P.; et al. Homeostatic PPARα Signaling Limits Inflammatory Responses to Commensal Microbiota in the Intestine. J. Immunol. 2016, 196, 4739–4749. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, L.; Mazzon, E.; Bruscoli, S.; Esposito, E.; Crisafulli, C.; Di Paola, R.; Caminiti, R.; Riccardi, C.; Cuzzocrea, S. Peroxisome proliferator-activated receptor-α modulates the anti-inflammatory effect of glucocorticoids in a model of inflammatory bowel disease in mice. Shock 2009, 31, 308–316. [Google Scholar] [CrossRef]
- Qi, Y.; Jiang, C.; Tanaka, N.; Krausz, K.W.; Brocker, C.N.; Fang, Z.-Z.; Bredell, B.X.; Shah, Y.M.; Gonzalez, F.J. PPARα-dependent exacerbation of experimental colitis by the hypolipidemic drug fenofibrate. Am. J. Physiol. Liver Physiol. 2014, 307, G564–G573. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Cao, L.; Jiang, C.; Xie, Y.; Cheng, X.; Krausz, K.W.; Qi, Y.; Sun, L.; Shah, Y.M.; Gonzalez, F.J.; et al. PPARα-UGT axis activation represses intestinal FXR-FGF15 feedback signalling and exacerbates experimental colitis. Nat. Commun. 2014, 5, 4573. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Song, Y.; Chai, Y.; Lu, F.; Gonzalez, F.J.; Fan, G.; Qi, Y. GC-MS metabolomics on PPARα-dependent exacerbation of colitis. Mol. BioSyst. 2015, 11, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Papamichael, K.; Lin, S.; Moore, M.; Papaioannou, G.; Sattler, L.; Cheifetz, A.S. Infliximab in inflammatory bowel disease. Ther. Adv. Chronic Dis. 2019, 10, 2040622319838443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ann, S.-J.; Chung, J.H.; Park, B.H.; Kim, S.H.; Jang, J.; Park, S.; Kang, S.-M.; Lee, S.-H. PPARα agonists inhibit inflammatory activation of macrophages through upregulation of β-defensin 1. Atherosclerosis 2015, 240, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Beisner, J.; Wang, G.; Nuding, S.; Oommen, S.T.; Kelly, D.; Parmentier-Decrucq, E.; Dessein, R.; Merour, E.; Chavatte, P.; et al. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc. Natl. Acad. Sci. USA 2010, 107, 8772–8777. [Google Scholar] [CrossRef] [Green Version]
- Muniz, L.R.; Knosp, C.; Yeretssian, G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front. Immunol. 2012, 3, 310. [Google Scholar] [CrossRef] [Green Version]
- Pentinmikko, N.; Iqbal, S.; Mana, M.; Andersson, S.; Cognetta, A.B., III; Suciu, R.M.; Roper, J.; Luopajärvi, K.; Markelin, E.; Gopalakrishnan, S.; et al. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nat. Cell Biol. 2019, 571, 398–402. [Google Scholar] [CrossRef]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef]
- Varnat, F.; Heggeler, B.B.-T.; Grisel, P.; Boucard, N.; Corthésy–Theulaz, I.; Wahli, W.; Desvergne, B. PPARβ/δ Regulates Paneth Cell Differentiation Via Controlling the Hedgehog Signaling Pathway. Gastroenterology 2006, 131, 538–553. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Raes, J.; van den Bogert, B.; Arumugam, M.; Booijink, C.C.G.M.; Troost, F.J.; Bork, P.; Wels, M.; De Vos, W.M.; Kleerebezem, M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012, 6, 1415–1426. [Google Scholar] [CrossRef]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; De Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.-Y.; Zhang, B.; He, W.-Q.; Zha, J.-M.; Odenwald, M.A.; Singh, G.; Tamura, A.; Shen, L.; Sailer, A.; Yeruva, S.; et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe 2017, 21, 671–681.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjöberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Denning, T.L. Segmented filamentous bacteria-induced immune responses: A balancing act between host protection and autoimmunity. Immunology 2018, 154, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedblom, G.A.; Reiland, H.A.; Sylte, M.J.; Johnson, T.J.; Baumler, D.J. Segmented Filamentous Bacteria—Metabolism Meets Immunity. Front. Microbiol. 2018, 9, 1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godlewski, G.; Offertáler, L.; Wagner, J.A.; Kunos, G. Receptors for acylethanolamides—GPR55 and GPR119. Prostaglandins Other Lipid Mediat. 2009, 89, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. J. Cereb. Blood Flow Metab. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Lauffer, L.M.; Iakoubov, R.; Brubaker, P.L. GPR119 Is Essential for Oleoylethanolamide-Induced Glucagon-Like Peptide-1 Secretion From the Intestinal Enteroendocrine L-Cell. Diabetes 2009, 58, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Petersen, G.; Sørensen, C.; Schmid, P.C.; Artmann, A.; Tang-Christensen, M.; Hansen, S.H.; Larsen, P.J.; Schmid, H.H.; Hansen, H.S. Intestinal levels of anandamide and oleoylethanolamide in food-deprived rats are regulated through their precursors. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2006, 1761, 143–150; discussion 141–142. [Google Scholar] [CrossRef]
- LoVerme, J.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The search for the palmitoylethanolamide receptor. Life Sci. 2005, 77, 1685–1698. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, M.; Iwasa, K.; Yoshikawa, K. Feeding regulation by oleoylethanolamide synthesized from dietary oleic acid. Prostaglandins Leukot. Essent. Fat. Acids 2020, 165, 102228. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, M.; Bonechi, E.; Provensi, G.; Costa, A.; Clarke, G.; Ballerini, C.; De Filippo, C.; Passani, M.B. Oleoylethanolamide treatment affects gut microbiota composition and the expression of intestinal cytokines in Peyer’s patches of mice. Sci. Rep. 2018, 8, 14881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial Ecology: Human Gut Microbes Associated with Obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The Gut Microbiota as an Environmental Factor That Regulates Fat Storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, C.; Liu, X.; Yang, J.; Feng, Y.; Wu, Y.; Xu, Y.; Zhu, Y.; Li, W. Fenofibrate Ameliorated Systemic and Retinal Inflammation and Modulated Gut Microbiota in High-Fat Diet-Induced Mice. Front. Cell Infect. Microbiol. 2022, 12, 839592. [Google Scholar] [CrossRef]
- Miller, T.L.; Wolin, M.J. Pathways of acetate, propionate, and butyrate formation by the human fecal microbial flora. Appl. Environ. Microbiol. 1996, 62, 1589–1592. [Google Scholar] [CrossRef] [Green Version]
- Parada Venegas, D.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-L.; Shu, C.-C.; Chen, Y.-M.; Lu, J.-J.; Wu, T.-S.; Lai, W.-F.; Tzeng, C.-M.; Lai, H.-C.; Lu, C.-C. Like Cures Like: Pharmacological Activity of Anti-Inflammatory Lipopolysaccharides From Gut Microbiome. Front. Pharmacol. 2020, 11, 554. [Google Scholar] [CrossRef]
- Vasques-Monteiro, I.M.L.; Silva-Veiga, F.M.; Miranda, C.S.; Gonçalves, C.B.D.A.; Daleprane, J.B.; Souza-Mello, V. A rise in Proteobacteria is an indicator of gut-liver axis-mediated nonalcoholic fatty liver disease in high-fructose-fed adult mice. Nutr. Res. 2021, 91, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bona, M.D.; Torres, C.H.d.M.; Lima, S.C.V.C.; Morais, A.H.D.A.; Lima, A.M.; Maciel, B.L.L. Intestinal Barrier Permeability in Obese Individuals with or without Metabolic Syndrome: A Systematic Review. Nutrients 2022, 14, 3649. [Google Scholar] [CrossRef] [PubMed]
- Veiga, F.M.S.; Miranda, C.S.; Martins, F.F.; Daleprane, J.B.; Mandarim-De-Lacerda, C.A.; Souza-Mello, V. Gut-liver axis modulation in fructose-fed mice: A role for PPAR-alpha and linagliptin. J. Endocrinol. 2020, 247, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Silva-Veiga, F.M.; Miranda, C.S.; Vasques-Monteiro, I.M.L.; Souza-Tavares, H.; Martins, F.F.; Daleprane, J.B.; Souza-Mello, V. Peroxisome proliferator-activated receptor-alpha activation and dipeptidyl peptidase-4 inhibition target dysbiosis to treat fatty liver in obese mice. World J. Gastroenterol. 2022, 28, 1814–1829. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Finan, B.; Bloom, S.; D’Alessio, D.; Drucker, D.; Flatt, P.; Fritsche, A.; Gribble, F.; Grill, H.; Habener, J.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Kirjavainen, P.V.; Ouwehand, A.C.; Isolauri, E.; Salminen, S.J. The ability of probiotic bacteria to bind to human intestinal mucus. FEMS Microbiol. Lett. 1998, 167, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Furrie, E.; Cummings, J.H.; Macfarlane, G.T. Chemotaxonomic Analysis of Bacterial Populations Colonizing the Rectal Mucosa in Patients with Ulcerative Colitis. Clin. Infect. Dis. 2004, 38, 1690–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cerbo, A.; Palmieri, B.; Aponte, M.; Morales-Medina, J.C.; Iannitti, T. Mechanisms and therapeutic effectiveness of lactobacilli. J. Clin. Pathol. 2016, 69, 187–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircalabioru, G.; Aviello, G.; Kubica, M.; Zhdanov, A.; Paclet, M.-H.; Brennan, L.; Hertzberger, R.; Papkovsky, D.; Bourke, B.; Knaus, U.G. Defensive Mutualism Rescues NADPH Oxidase Inactivation in Gut Infection. Cell Host Microbe 2016, 19, 651–663. [Google Scholar] [CrossRef]
- Singh, A.K.; Hertzberger, R.Y.; Knaus, U.G. Hydrogen peroxide production by lactobacilli promotes epithelial restitution during colitis. Redox Biol. 2018, 16, 11–20. [Google Scholar] [CrossRef]
- Goto, T.; Kim, Y.-I.; Furuzono, T.; Takahashi, N.; Yamakuni, K.; Yang, H.-E.; Li, Y.; Ohue, R.; Nomura, W.; Sugawara, T.; et al. 10-oxo-12(Z)-octadecenoic acid, a linoleic acid metabolite produced by gut lactic acid bacteria, potently activates PPARγ and stimulates adipogenesis. Biochem. Biophys. Res. Commun. 2015, 459, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Nagatake, T.; Kishino, S.; Urano, E.; Murakami, H.; Kitamura, N.; Konishi, K.; Ohno, H.; Tiwari, P.; Morimoto, S.; Node, E.; et al. Intestinal microbe-dependent ω3 lipid metabolite αKetoA prevents inflammatory diseases in mice and cynomolgus macaques. Mucosal Immunol. 2022, 15, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Kishino, S.; Takeuchi, M.; Park, S.-B.; Hirata, A.; Kitamura, N.; Kunisawa, J.; Kiyono, H.; Iwamoto, R.; Isobe, Y.; Arita, M.; et al. Polyunsaturated fatty acid saturation by gut lactic acid bacteria affecting host lipid composition. Proc. Natl. Acad. Sci. USA 2013, 110, 17808–17813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.A.P.; Harris, H.M.B.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Miyamoto, J.; Mizukure, T.; Park, S.-B.; Kishino, S.; Kimura, I.; Hirano, K.; Bergamo, P.; Rossi, M.; Suzuki, T.; Arita, M.; et al. A Gut Microbial Metabolite of Linoleic Acid, 10-Hydroxy-cis-12-octadecenoic Acid, Ameliorates Intestinal Epithelial Barrier Impairment Partially via GPR40-MEK-ERK Pathway. J. Biol. Chem. 2015, 290, 2902–2918. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-H.; Jeong, D.; Kang, I.-B.; Kim, H.; Song, K.-Y.; Seo, K.-H. Dual function of Lactobacillus kefiri DH5 in preventing high-fat-diet-induced obesity: Direct reduction of cholesterol and upregulation of PPAR-α in adipose tissue. Mol. Nutr. Food Res. 2017, 61, 1700252. [Google Scholar] [CrossRef]
- Nakamura, F.; Ishida, Y.; Aihara, K.; Sawada, D.; Ashida, N.; Sugawara, T.; Aoki, Y.; Takehara, I.; Takano, K.; Fujiwara, S. Effect of fragmented Lactobacillus amylovorus CP1563 on lipid metabolism in overweight and mildly obese individuals: A randomized controlled trial. Microb. Ecol. Health Dis. 2016, 27, 30312. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, F.; Ishida, Y.; Sawada, D.; Ashida, N.; Sugawara, T.; Sakai, M.; Goto, T.; Kawada, T.; Fujiwara, S. Fragmented Lactic Acid Bacterial Cells Activate Peroxisome Proliferator-Activated Receptors and Ameliorate Dyslipidemia in Obese Mice. J. Agric. Food Chem. 2016, 64, 2549–2559. [Google Scholar] [CrossRef]
- Cai, H.; Wen, Z.; Li, X.; Meng, K.; Yang, P. Lactobacillus plantarum FRT10 alleviated high-fat diet–induced obesity in mice through regulating the PPARα signal pathway and gut microbiota. Appl. Microbiol. Biotechnol. 2020, 104, 5959–5972. [Google Scholar] [CrossRef]
- Jiang, J.; Xiong, J.; Ni, J.; Chen, C.; Wang, K. Live Combined B. subtilis and E. faecium Alleviate Liver Inflammation, Improve Intestinal Barrier Function, and Modulate Gut Microbiota in Mice with Non-Alcoholic Fatty Liver Disease. Med. Sci. Monit. 2021, 27, 931143. [Google Scholar] [CrossRef]
- Yang, J.-Y.; Lee, Y.-S.; Kim, Y.; Lee, S.-H.; Ryu, S.; Fukuda, S.; Hase, K.; Yang, C.-S.; Lim, H.S.; Kim, M.-S.; et al. Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal Immunol. 2017, 10, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, S.M.; Sepahi, A.A.; Mousavi, S.F.; Vaziri, F.; Siadat, S.D. The effect of Faecalibacterium prausnitzii and its extracellular vesicles on the permeability of intestinal epithelial cells and expression of PPARs and ANGPTL4 in the Caco-2 cell culture model. J. Diabetes Metab. Disord. 2020, 19, 1061–1069. [Google Scholar] [CrossRef]
- Deleu, S.; Machiels, K.; Raes, J.; Verbeke, K.; Vermeire, S. Short chain fatty acids and its producing organisms: An overlooked therapy for IBD? eBioMedicine 2021, 66, 103293. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Q.; Yang, Y.; Guo, A. Biological Function of Short-Chain Fatty Acids and Its Regulation on Intestinal Health of Poultry. Front. Veter Sci. 2021, 8, 736739. [Google Scholar] [CrossRef]
- Higashimura, Y.; Naito, Y.; Takagi, T.; Uchiyama, K.; Mizushima, K.; Yoshikawa, T. Propionate Promotes Fatty Acid Oxidation through the Up-Regulation of Peroxisome Proliferator-Activated Receptor α in Intestinal Epithelial Cells. J. Nutr. Sci. Vitaminol. 2015, 61, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Kasubuchi, M.; Hasegawa, S.; Hiramatsu, T.; Ichimura, A.; Kimura, I. Dietary Gut Microbial Metabolites, Short-chain Fatty Acids, and Host Metabolic Regulation. Nutrients 2015, 7, 2839–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhang, M.; Wang, Y.; Dorfman, R.G.; Liu, H.; Yu, T.; Chen, X.; Tang, D.; Xu, L.; Yin, Y.; et al. Faecalibacterium prausnitzii Produces Butyrate to Maintain Th17/Treg Balance and to Ameliorate Colorectal Colitis by Inhibiting Histone Deacetylase 1. Inflamm. Bowel Dis. 2018, 24, 1926–1940. [Google Scholar] [CrossRef] [Green Version]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef]
- Martinez-Medina, M.; Aldeguer, X.; Gonzalez-Huix, F.; Acero, D.; Garcia-Gil, J.L. Abnormal microbiota composition in the ileocolonic mucosa of Crohnʼs disease patients as revealed by polymerase chain reaction-denaturing gradient gel electrophoresis. Inflamm. Bowel Dis. 2006, 12, 1136–1145. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, M.C.; Derrien, M.; Isolauri, E.; de Vos, W.M.; Salminen, S. Intestinal Integrity and Akkermansia muciniphila, a Mucin-Degrading Member of the Intestinal Microbiota Present in Infants, Adults, and the Elderly. Appl. Environ. Microbiol. 2007, 73, 7767–7770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Müller, M.; de Vos, W.M. Modulation of Mucosal Immune Response, Tolerance, and Proliferation in Mice Colonized by the Mucin-Degrader Akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reunanen, J.; Kainulainen, V.; Huuskonen, L.; Ottman, N.; Belzer, C.; Huhtinen, H.; de Vos, W.M.; Satokari, R. Akkermansia muciniphila Adheres to Enterocytes and Strengthens the Integrity of the Epithelial Cell Layer. Appl. Environ. Microbiol. 2015, 81, 3655–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelakkot, C.; Choi, Y.; Kim, D.-K.; Park, H.T.; Ghim, J.; Kwon, Y.; Jeon, J.; Kim, M.-S.; Jee, Y.-K.; Gho, Y.S.; et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp. Mol. Med. 2018, 50, e450. [Google Scholar] [CrossRef] [Green Version]
- Ashrafian, F.; Behrouzi, A.; Shahriary, A.; Badi, S.A.; Davari, M.; Khatami, S.; Jamnani, F.R.; Fateh, A.; Vaziri, F.; Siadat, S.D. Comparative study of effect of Akkermansia muciniphila and its extracellular vesicles on toll-like receptors and tight junction. Gastroenterol. Hepatol. Bed Bench 2019, 12, 163–168. [Google Scholar]
- Liu, Y.; Yang, M.; Tang, L.; Wang, F.; Huang, S.; Liu, S.; Lei, Y.; Wang, S.; Xie, Z.; Wang, W.; et al. TLR4 regulates RORγt+ regulatory T-cell responses and susceptibility to colon inflammation through interaction with Akkermansia muciniphila. Microbiome 2022, 10, 98. [Google Scholar] [CrossRef]
- Earley, H.; Lennon, G.; Balfe, A.; Coffey, J.C.; Winter, D.C.; O’Connell, P.R. The abundance of Akkermansia muciniphila and its relationship with sulphated colonic mucins in health and ulcerative colitis. Sci. Rep. 2019, 9, 15683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Ji, X.; Lu, G.; Zhang, F. The potential of Akkermansia muciniphila in inflammatory bowel disease. Appl. Microbiol. Biotechnol. 2021, 105, 5785–5794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, P.; Wu, X.; Lu, G.; Marcella, C.; Ji, X.; Ji, G.; Zhang, F. Alterations of Akkermansia muciniphila in the inflammatory bowel disease patients with washed microbiota transplantation. Appl. Microbiol. Biotechnol. 2020, 104, 10203–10215. [Google Scholar] [CrossRef] [PubMed]
- Depommier, C.; Vitale, R.M.; Iannotti, F.A.; Silvestri, C.; Flamand, N.; Druart, C.; Everard, A.; Pelicaen, R.; Maiter, D.; Thissen, J.-P.; et al. Beneficial Effects of Akkermansia muciniphila Are Not Associated with Major Changes in the Circulating Endocannabinoidome but Linked to Higher Mono-Palmitoyl-Glycerol Levels as New PPARα Agonists. Cells 2021, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crakes, K.R.; Rocha, C.S.; Grishina, I.; Hirao, L.A.; Napoli, E.; Gaulke, C.A.; Fenton, A.; Datta, S.; Arredondo, J.; Marco, M.L.; et al. PPARα-targeted mitochondrial bioenergetics mediate repair of intestinal barriers at the host–microbe intersection during SIV infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24819–24829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depommier, C.; Van Hul, M.; Everard, A.; Delzenne, N.M.; De Vos, W.M.; Cani, P.D. Pasteurized Akkermansia muciniphila increases whole-body energy expenditure and fecal energy excretion in diet-induced obese mice. Gut Microbes 2020, 11, 1231–1245. [Google Scholar] [CrossRef] [Green Version]
- Payahoo, L.; Khajebishak, Y.; Alivand, M.R.; Soleimanzadeh, H.; Alipour, S.; Barzegari, A.; Ostadrahimi, A. Investigation the effect of oleoylethanolamide supplementation on the abundance of Akkermansia muciniphila bacterium and the dietary intakes in people with obesity: A randomized clinical trial. Appetite 2019, 141, 104301. [Google Scholar] [CrossRef]
- Crawford, P.A.; Crowley, J.R.; Sambandam, N.; Muegge, B.D.; Costello, E.K.; Hamady, M.; Knight, R.; Gordon, J.I. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 11276–11281. [Google Scholar] [CrossRef] [Green Version]
- Tiso, M.; Schechter, A.N. Nitrate Reduction to Nitrite, Nitric Oxide and Ammonia by Gut Bacteria under Physiological Conditions. PLoS ONE 2015, 10, e0119712. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.; Ferreira, N.R.; Rocha, B.S.; Barbosa, R.M.; Laranjinha, J. The redox interplay between nitrite and nitric oxide: From the gut to the brain. Redox Biol. 2013, 1, 276–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parham, N.J.; Gibson, G.R. Microbes involved in dissimilatory nitrate reduction in the human large intestine. FEMS Microbiol. Ecol. 2000, 31, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Vermeiren, J.; Van de Wiele, T.; Verstraete, W.; Boeckx, P.; Boon, N. Nitric Oxide Production by the Human Intestinal Microbiota by Dissimilatory Nitrate Reduction to Ammonium. J. Biomed. Biotechnol. 2009, 2009, 284718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumft, W.G. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.; Cooper, C.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Sanders, K.M.; Ward, S.M. Nitric oxide and its role as a non-adrenergic, non-cholinergic inhibitory neurotransmitter in the gastrointestinal tract. Br. J. Pharmacol. 2019, 176, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.-O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Pittman, R.N.; Popel, A. Nitric Oxide in the Vasculature: Where Does It Come From and Where Does It Go? A Quantitative Perspective. Antioxidants Redox Signal. 2008, 10, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [Green Version]
- Forrester, M.T.; Foster, M.W. Protection from nitrosative stress: A central role for microbial flavohemoglobin. Free Radic. Biol. Med. 2012, 52, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Oguchi, H.; Kobayashi, T.; Kusama, S.; Sugiura, R.; Moriya, K.; Hirata, T.; Yukioka, Y.; Takaya, N.; Yajima, S.; et al. Nitrogen oxide cycle regulates nitric oxide levels and bacterial cell signaling. Sci. Rep. 2016, 6, 22038. [Google Scholar] [CrossRef] [PubMed]
- Cookson, T.A. Bacterial-Induced Blood Pressure Reduction: Mechanisms for the Treatment of Hypertension via the Gut. Front. Cardiovasc. Med. 2021, 8, 721393. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.S.; Tribble, G.; Angelov, N. Oral Microbiome and Nitric Oxide: The Missing Link in the Management of Blood Pressure. Curr. Hypertens. Rep. 2017, 19, 33. [Google Scholar] [CrossRef] [PubMed]
- Spinas, G.A. The Dual Role of Nitric Oxide in Islet β-Cells. Physiology 1999, 14, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Raithel, M.; Hagel, A.F.; Zopf, Y.; Bijlsma, P.B.; De Rossi, T.M.; Gabriel, S.; Weidenhiller, M.; Kressel, J.; Hahn, E.G.; Konturek, P.C. Analysis of immediate ex vivo release of nitric oxide from human colonic mucosa in gastrointestinally mediated allergy, inflammatory bowel disease and controls. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2012, 63, 317–325. [Google Scholar]
- Taheri, P.; Mohammadi, F.; Nazeri, M.; Zarei, M.R.; Chamani, G.; Esfahlani, M.A.; Taheri, F.; Shabani, M. Nitric oxide role in anxiety-like behavior, memory and cognitive impairments in animal model of chronic migraine. Heliyon 2020, 6, e05654. [Google Scholar] [CrossRef] [PubMed]
- Joca, S.R.L.; Sartim, A.G.; Roncalho, A.L.; Diniz, C.F.A.; Wegener, G. Nitric oxide signalling and antidepressant action revisited. Cell Tissue Res. 2019, 377, 45–58. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Liu, Y.; Wang, Y.; Fan, R.; Hu, X.; Zhang, F.; Yang, J.; Chen, J. The Role of Intestinal Mucosal Barrier in Autoimmune Disease: A Potential Target. Front. Immunol. 2022, 13, 871713. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. All disease begins in the (leaky) gut: Role of zonulin-mediated gut permeability in the pathogenesis of some chronic inflammatory diseases. F1000Research 2020, 9, 69. [Google Scholar] [CrossRef]
- Kaminsky, L.W.; Al-Sadi, R.; Ma, T.Y. IL-1β and the Intestinal Epithelial Tight Junction Barrier. Front. Immunol. 2021, 12, 767456. [Google Scholar] [CrossRef] [PubMed]
- Ngo, P.A.; Neurath, M.F.; López-Posadas, R. Impact of Epithelial Cell Shedding on Intestinal Homeostasis. Int. J. Mol. Sci. 2022, 23, 4160. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.M.; Campbell, B.J.; Pritchard, D.M. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Glover, L.E.; Lee, J.S.; Colgan, S.P. Oxygen metabolism and barrier regulation in the intestinal mucosa. J. Clin. Investig. 2016, 126, 3680–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; Bayless, A.J.; Kominsky, D.J.; et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol. Biol. Cell 2015, 26, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karwad, M.; Couch, D.; Wright, K.; Tufarelli, C.; Larvin, M.; Lund, J.; O’Sullivan, S. Endocannabinoids and endocannabinoid-like compounds modulate hypoxia-induced permeability in CaCo-2 cells via CB1, TRPV1, and PPARα. Biochem. Pharmacol. 2019, 168, 465–472. [Google Scholar] [CrossRef]
- Mazzon, E.; Cuzzocrea, S. Absence of functional peroxisome proliferator-activated receptor-α enhanced ileum permeability during experimental colitis. Shock 2007, 28, 192–201. [Google Scholar] [CrossRef]
- Mazzon, E.; Cuzzocrea, S. Role of TNF-α in ileum tight junction alteration in mouse model of restraint stress. Am. J. Physiol. Liver Physiol. 2008, 294, G1268–G1280. [Google Scholar] [CrossRef]
- Fukui, H. Increased Intestinal Permeability and Decreased Barrier Function: Does It Really Influence the Risk of Inflammation? Inflamm. Intest. Dis. 2016, 1, 135–145. [Google Scholar] [CrossRef]
- Crakes, K.R.; Pires, J.; Quach, N.; Ellis-Reis, R.E.; Greathouse, R.; Chittum, K.A.; Steiner, J.M.; Pesavento, P.; Marks, S.L.; Dandekar, S.; et al. Fenofibrate promotes PPARα-targeted recovery of the intestinal epithelial barrier at the host-microbe interface in dogs with diabetes mellitus. Sci. Rep. 2021, 11, 13454. [Google Scholar] [CrossRef]
- Lama, A.; Provensi, G.; Amoriello, R.; Pirozzi, C.; Rani, B.; Mollica, M.P.; Raso, G.M.; Ballerini, C.; Meli, R.; Passani, M.B. The anti-inflammatory and immune-modulatory effects of OEA limit DSS-induced colitis in mice. Biomed. Pharmacother. 2020, 129, 110368. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Shi, Y.; Yuan, J.; Sa, R.; Chen, W.; Wan, X. Matrine protects against DSS-induced murine colitis by improving gut barrier integrity, inhibiting the PPAR-α signaling pathway, and modulating gut microbiota. Int. Immunopharmacol. 2021, 100, 108091. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; Van De Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Ser. Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Narravula, S.; Colgan, S.P. Hypoxia-Inducible Factor 1-Mediated Inhibition of Peroxisome Proliferator-Activated Receptor α Expression During Hypoxia. J. Immunol. 2001, 166, 7543–7548. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Moran, E.; Ding, L.; Cheng, R.; Xu, X.; Ma, J.-X. PPARα Regulates Mobilization and Homing of Endothelial Progenitor Cells Through the HIF-1α/SDF-1 Pathway. Investig. Opthalmology Vis. Sci. 2014, 55, 3820–3832. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhang, S.; Xue, J.; Avery, J.; Wu, J.; Lind, S.E.; Ding, W.-Q. Activation of Peroxisome Proliferator-activated Receptor α (PPARα) Suppresses Hypoxia-inducible Factor-1α (HIF-1α) Signaling in Cancer Cells. J. Biol. Chem. 2012, 287, 35161–35169. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.B.; Colorado, I.; Reino, D.; Palange, D.; Lu, Q.; Qin, X.; Abungu, B.; Watkins, A.; Caputo, F.J.; Xu, D.-Z.; et al. Hypoxia-inducible factor plays a gut-injurious role in intestinal ischemia reperfusion injury. Am. J. Physiol. Liver Physiol. 2011, 300, G853–G861. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Pignataro, G.; Molinaro, P.; Irace, C.; Scorziello, A.; Di Renzo, G.F.; Annunziato, L. HIF-1alpha reveals a binding activity to the promoter of iNOS gene after permanent middle cerebral artery occlusion. J. Neurochem. 2004, 90, 368–378. [Google Scholar] [CrossRef]
- Chowdhury, R.; Godoy, L.C.; Thiantanawat, A.; Trudel, L.J.; Deen, W.M.; Wogan, G.N. Nitric Oxide Produced Endogenously Is Responsible for Hypoxia-Induced HIF-1α Stabilization in Colon Carcinoma Cells. Chem. Res. Toxicol. 2012, 25, 2194–2202. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yin, L.-H.; Gao, M.; Xu, L.-N.; Qi, Y.; Peng, J.-Y. MiR-23a-5p exacerbates intestinal ischemia–reperfusion injury by promoting oxidative stress via targeting PPAR alpha. Biochem. Pharmacol. 2020, 180, 114194. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Yan, Q.; Wang, Z.; Liu, D.; Zhan, M.; Du, J.; Chen, L. Oleoylethanolamide Alleviates Hepatic Ischemia-Reperfusion Injury via Inhibiting Endoplasmic Reticulum Stress-Associated Apoptosis. PPAR Res. 2022, 2022, 2212996. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabacka, M.; Płonka, P.M.; Pierzchalska, M. The PPARα Regulation of the Gut Physiology in Regard to Interaction with Microbiota, Intestinal Immunity, Metabolism, and Permeability. Int. J. Mol. Sci. 2022, 23, 14156. https://doi.org/10.3390/ijms232214156
Grabacka M, Płonka PM, Pierzchalska M. The PPARα Regulation of the Gut Physiology in Regard to Interaction with Microbiota, Intestinal Immunity, Metabolism, and Permeability. International Journal of Molecular Sciences. 2022; 23(22):14156. https://doi.org/10.3390/ijms232214156
Chicago/Turabian StyleGrabacka, Maja, Przemysław M. Płonka, and Małgorzata Pierzchalska. 2022. "The PPARα Regulation of the Gut Physiology in Regard to Interaction with Microbiota, Intestinal Immunity, Metabolism, and Permeability" International Journal of Molecular Sciences 23, no. 22: 14156. https://doi.org/10.3390/ijms232214156