
Adaptive Immunity to Viruses: What Did We Learn from SARS-CoV-2 Infection?
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The First Events after SARS-CoV-2 Infection
3. Characteristics of Adaptive Immunity in COVID-19
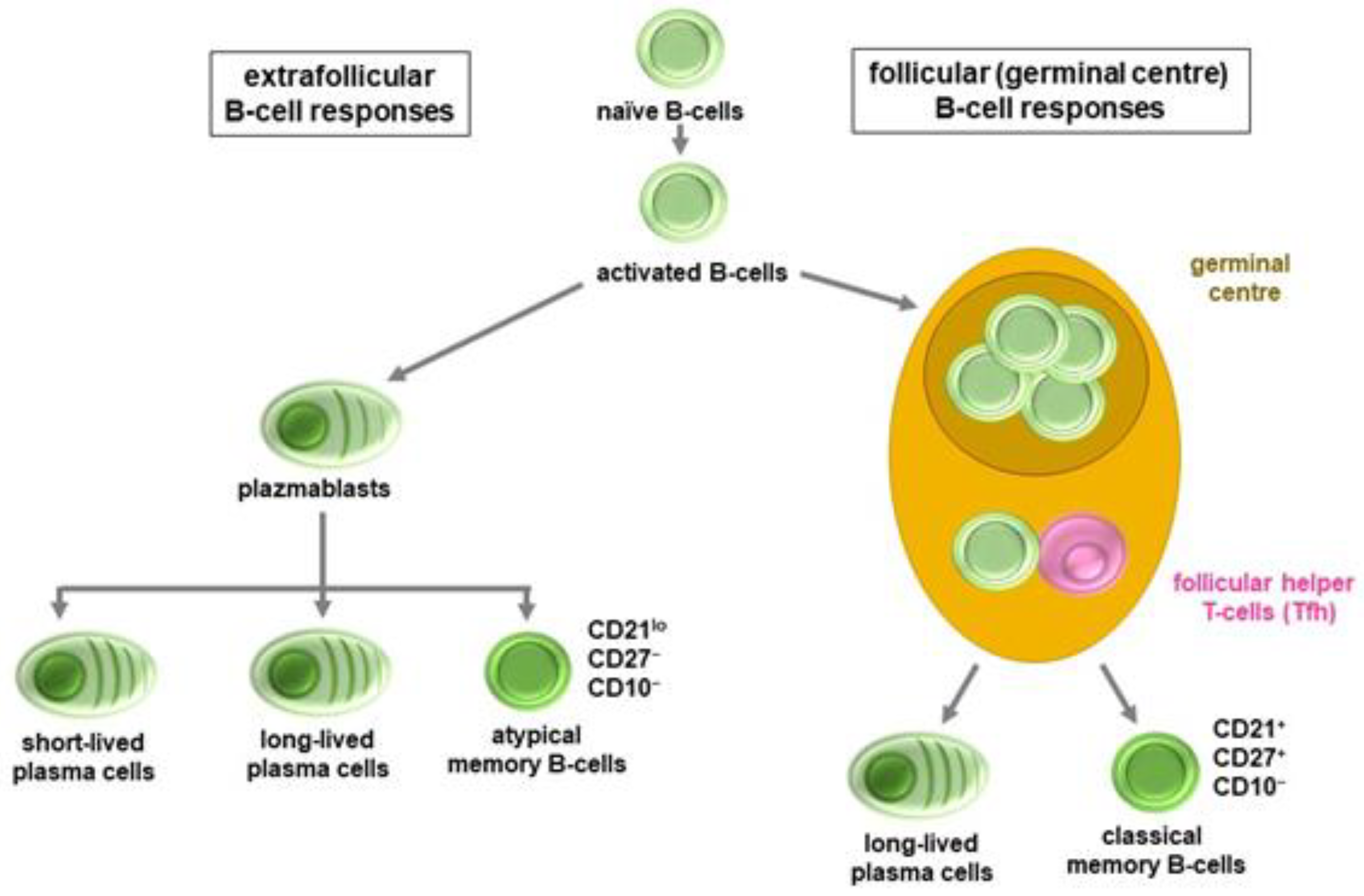
3.1. B-Cell and Antibody Responses
3.2. T-Cell Responses
3.3. Immunological Memory in COVID-19
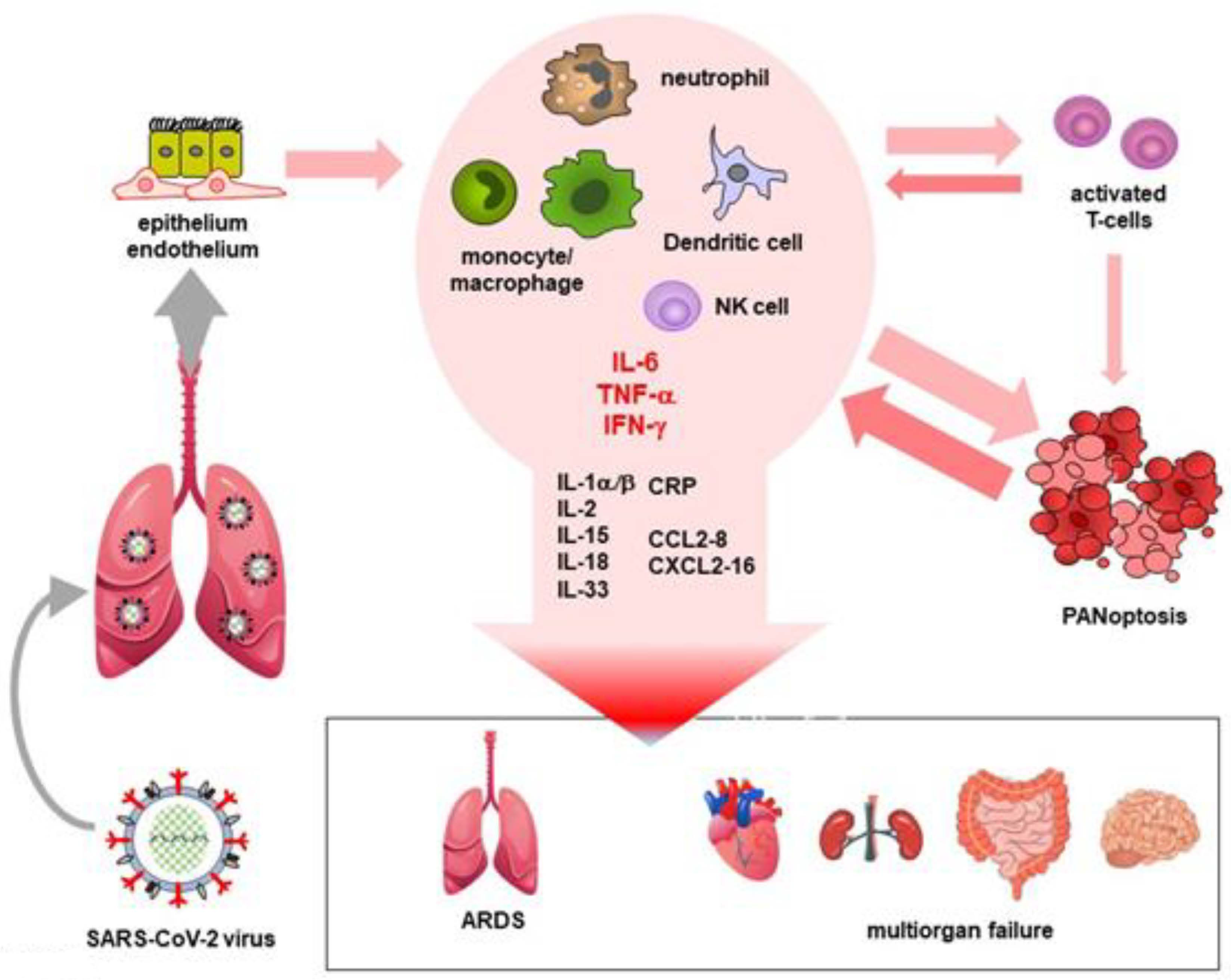
4. Cytokine Storm and MIS
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S.; Rajagopal, S.; Pai, A.R.; Kutty, S. Cytokine Storm in COVID-19-Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; He, L.; Zhang, Q.; Huang, Z.; Che, X.; Hou, J.; Wang, H.; Shen, H.; Qiu, L.; Li, Z.; et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J. Pathol. 2004, 203, 622–630. [Google Scholar] [CrossRef]
- Ruiz-Galiana, J.; De Lucas Ramos, P.; Garcia-Botella, A.; Garcia-Lledo, A.; Gomez-Pavon, J.; Gonzalez Del Castillo, J.; Hernandez-Sampelayo, T.; Martin-Delgado, M.C.; Martin Sanchez, F.J.; Martinez-Selles, M.; et al. Persistence and viability of SARS-CoV-2 in primary infection and reinfections. Rev. Esp. Quimioter. 2022, 35, 1–6. [Google Scholar] [CrossRef]
- Martinez-Chinchilla, C.; Vazquez-Montero, L.; Palazon-Carrion, N.; Fernandez-Roman, I.M.; Lopez-Barba, J.; de la Cruz-Merino, L.; Rodriguez-Bano, J.; Palacios-Baena, Z.R. Persistence of SARS-CoV-2 Infection in Severely Immunocompromised Patients with Complete Remission B-Cell Lymphoma and Anti-CD20 Monoclonal Antibody Therapy: A Case Report of Two Cases. Front. Immunol. 2022, 13, 860891. [Google Scholar] [CrossRef]
- Wong, L.R.; Perlman, S. Immune dysregulation and immunopathology induced by SARS-CoV-2 and related coronaviruses—Are we our own worst enemy? Nat. Rev. Immunol. 2022, 22, 47–56. [Google Scholar] [CrossRef]
- Uher, F.; Matula, Z.; Gonczi, M.; Gopcsa, L.; Beko, G.; Reti, M.; Szekanecz, Z.; Ajzner, E.; Valyi-Nagy, I. Immune response against the severe acute respiratory syndrome coronavirus 2. Orv. Hetil. 2022, 163, 774–787. [Google Scholar] [CrossRef]
- Lee, S.; Channappanavar, R.; Kanneganti, T.D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020, 41, 1083–1099. [Google Scholar] [CrossRef]
- Gierer, S.; Bertram, S.; Kaup, F.; Wrensch, F.; Heurich, A.; Kramer-Kuhl, A.; Welsch, K.; Winkler, M.; Meyer, B.; Drosten, C.; et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 2013, 87, 5502–5511. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.; Krammer, F.; Iwasaki, A. The first 12 months of COVID-19: A timeline of immunological insights. Nat. Rev. Immunol. 2021, 21, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Thieme, C.J.; Anft, M.; Paniskaki, K.; Blazquez-Navarro, A.; Doevelaar, A.; Seibert, F.S.; Hoelzer, B.; Konik, M.J.; Berger, M.M.; Brenner, T.; et al. Robust T Cell Response toward Spike, Membrane, and Nucleocapsid SARS-CoV-2 Proteins Is Not Associated with Recovery in Critical COVID-19 Patients. Cell Rep. Med. 2020, 1, 100092. [Google Scholar] [CrossRef]
- Galipeau, Y.; Greig, M.; Liu, G.; Driedger, M.; Langlois, M.A. Humoral Responses and Serological Assays in SARS-CoV-2 Infections. Front. Immunol. 2020, 11, 610688. [Google Scholar] [CrossRef]
- Post, N.; Eddy, D.; Huntley, C.; van Schalkwyk, M.C.I.; Shrotri, M.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P.; et al. Antibody response to SARS-CoV-2 infection in humans: A systematic review. PLoS ONE 2020, 15, e0244126. [Google Scholar] [CrossRef]
- Hoepel, W.; Chen, H.J.; Geyer, C.E.; Allahverdiyeva, S.; Manz, X.D.; de Taeye, S.W.; Aman, J.; Mes, L.; Steenhuis, M.; Griffith, G.R.; et al. High titers and low fucosylation of early human anti-SARS-CoV-2 IgG promote inflammation by alveolar macrophages. Sci. Transl. Med. 2021, 13, eabf8654. [Google Scholar] [CrossRef]
- Ullah, I.; Prevost, J.; Ladinsky, M.S.; Stone, H.; Lu, M.; Anand, S.P.; Beaudoin-Bussieres, G.; Symmes, K.; Benlarbi, M.; Ding, S.; et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity 2021, 54, 2143–2158.e5. [Google Scholar] [CrossRef]
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, M.C.; Ramonell, R.P.; Nguyen, D.C.; Cashman, K.S.; Saini, A.S.; Haddad, N.S.; Ley, A.M.; Kyu, S.; Howell, J.C.; Ozturk, T.; et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 2020, 21, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Ellebedy, A.H. The germinal centre B cell response to SARS-CoV-2. Nat. Rev. Immunol. 2022, 22, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. Covid-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar] [CrossRef]
- Halpert, G.; Shoenfeld, Y. SARS-CoV-2, the autoimmune virus. Autoimmun. Rev. 2020, 19, 102695. [Google Scholar] [CrossRef]
- Knight, J.S.; Caricchio, R.; Casanova, J.L.; Combes, A.J.; Diamond, B.; Fox, S.E.; Hanauer, D.A.; James, J.A.; Kanthi, Y.; Ladd, V.; et al. The intersection of COVID-19 and autoimmunity. J. Clin. Investig. 2021, 131, e154886. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Chao, Y.X.; Rotzschke, O.; Tan, E.K. The role of IgA in COVID-19. Brain Behav. Immun. 2020, 87, 182–183. [Google Scholar] [CrossRef]
- Pierce, C.A.; Sy, S.; Galen, B.; Goldstein, D.Y.; Orner, E.; Keller, M.J.; Herold, K.C.; Herold, B.C. Natural mucosal barriers and COVID-19 in children. JCI Insight 2021, 6, e148694. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.J.; Peltan, I.D.; Jensen, P.; Hoda, D.; Hunter, B.; Silver, A.; Starr, N.; Buckel, W.; Grisel, N.; Hummel, E.; et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: A cohort study. Lancet Rheumatol. 2020, 2, e754–e763. [Google Scholar] [CrossRef]
- Jarjour, N.N.; Masopust, D.; Jameson, S.C. T Cell Memory: Understanding COVID-19. Immunity 2021, 54, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Niessl, J.; Sekine, T.; Buggert, M. T cell immunity to SARS-CoV-2. Semin. Immunol. 2021, 55, 101505. [Google Scholar] [CrossRef]
- Jergovic, M.; Coplen, C.P.; Uhrlaub, J.L.; Nikolich-Zugich, J. Immune response to COVID-19 in older adults. J. Heart Lung Transplant. 2021, 40, 1082–1089. [Google Scholar] [CrossRef]
- Grifoni, A.; Sidney, J.; Vita, R.; Peters, B.; Crotty, S.; Weiskopf, D.; Sette, A. SARS-CoV-2 human T cell epitopes: Adaptive immune response against COVID-19. Cell Host Microbe 2021, 29, 1076–1092. [Google Scholar] [CrossRef]
- Keeton, R.; Tincho, M.B.; Ngomti, A.; Baguma, R.; Benede, N.; Suzuki, A.; Khan, K.; Cele, S.; Bernstein, M.; Karim, F.; et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 2022, 603, 488–492. [Google Scholar] [CrossRef]
- Bjorkstrom, N.K.; Ponzetta, A. Natural killer cells and unconventional T cells in COVID-19. Curr. Opin. Virol. 2021, 49, 176–182. [Google Scholar] [CrossRef]
- Orumaa, K.; Dunne, M.R. The role of unconventional T cells in COVID-19. Ir. J. Med. Sci. 2022, 191, 519–528. [Google Scholar] [CrossRef]
- Siggins, M.K.; Thwaites, R.S.; Openshaw, P.J.M. Durability of Immunity to SARS-CoV-2 and Other Respiratory Viruses. Trends Microbiol. 2021, 29, 648–662. [Google Scholar] [CrossRef]
- White, H.N. B-Cell Memory Responses to Variant Viral Antigens. Viruses 2021, 13, 565. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 2021, 371, eabf4063. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Huang, Z.T.; Li, L.; Wu, M.H.; Yu, T.; Koup, R.A.; Bailer, R.T.; Wu, C.Y. Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch. Virol. 2009, 154, 1093–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zheng, Y.; Zhao, L.; Yue, Z.; Pan, F.; Chen, Y.; Yu, B.; Chen, Y.; Zhao, G.; Zhou, Y.; et al. Investigation of the impact of SARS-CoV infection on the immunologic status and lung function after 15 years. BMC Infect. Dis. 2021, 21, 1183. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Crotty, S. Pre-existing immunity to SARS-CoV-2: The knowns and unknowns. Nat. Rev. Immunol. 2020, 20, 457–458. [Google Scholar] [CrossRef]
- Willyard, C. What the Omicron wave is revealing about human immunity. Nature 2022, 602, 22–25. [Google Scholar] [CrossRef]
- Cohen, K.W.; Linderman, S.L.; Moodie, Z.; Czartoski, J.; Lai, L.; Mantus, G.; Norwood, C.; Nyhoff, L.E.; Edara, V.V.; Floyd, K.; et al. Longitudinal analysis shows durable and broad immune memory after SARS-CoV-2 infection with persisting antibody responses and memory B and T cells. Cell Rep. Med. 2021, 2, 100354. [Google Scholar] [CrossRef]
- Kolehmainen, P.; Heroum, J.; Jalkanen, P.; Huttunen, M.; Toivonen, L.; Marjomaki, V.; Waris, M.; Smura, T.; Kakkola, L.; Tauriainen, S.; et al. Serological Follow-Up Study Indicates High Seasonal Coronavirus Infection and Reinfection Rates in Early Childhood. Microbiol. Spectr. 2022, 10, e0196721. [Google Scholar] [CrossRef]
- Meyerholz, D.K.; Perlman, S. Does common cold coronavirus infection protect against severe SARS-CoV-2 disease? J. Clin. Investig. 2021, 131, e144807. [Google Scholar] [CrossRef]
- Kundu, R.; Narean, J.S.; Wang, L.; Fenn, J.; Pillay, T.; Fernandez, N.D.; Conibear, E.; Koycheva, A.; Davies, M.; Tolosa-Wright, M.; et al. Cross-reactive memory T cells associate with protection against SARS-CoV-2 infection in COVID-19 contacts. Nat. Commun. 2022, 13, 80. [Google Scholar] [CrossRef]
- Aggarwal, A.; Stella, A.O.; Walker, G.; Akerman, A.; Esneau, C.; Milogiannakis, V.; Burnett, D.L.; McAllery, S.; Silva, M.R.; Lu, Y.; et al. Platform for isolation and characterization of SARS-CoV-2 variants enables rapid characterization of Omicron in Australia. Nat. Microbiol. 2022, 7, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Cai, C.; Grifoni, A.; Muller, T.R.; Niessl, J.; Olofsson, A.; Humbert, M.; Hansson, L.; Osterborg, A.; Bergman, P.; et al. Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat. Med. 2022, 28, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Lynch, J.B.; Del Rio, C. Mild or Moderate COVID-19. N. Engl. J. Med. 2020, 383, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K.; Mehra, M.R. COVID-19 illness in native and immunosuppressed states: A clinical-therapeutic staging proposal. J. Heart Lung Transplant. 2020, 39, 405–407. [Google Scholar] [CrossRef] [Green Version]
- Szekanecz, Z.; Bogos, K.; Constantin, T.; Fulesdi, B.; Muller, V.; Rakoczi, E.; Varkonyi, I.; Valyi-Nagy, I. Antiviral and anti-inflammatory therapies in COVID-19. Hung Med. J. 2021, 162, 643–651. [Google Scholar] [CrossRef]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9, e59177. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Merrill, J.T.; Erkan, D.; Winakur, J.; James, J.A. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat. Rev. Rheumatol. 2020, 16, 581–589. [Google Scholar] [CrossRef]
- Borges, L.; Pithon-Curi, T.C.; Curi, R.; Hatanaka, E. COVID-19 and Neutrophils: The Relationship between Hyperinflammation and Neutrophil Extracellular Traps. Mediat. Inflamm. 2020, 2020, 8829674. [Google Scholar] [CrossRef]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2020, 93, 250–256. [Google Scholar] [CrossRef]
- Caricchio, R.; Gallucci, M.; Dass, C.; Zhang, X.; Gallucci, S.; Fleece, D.; Bromberg, M.; Criner, G.J.; Temple University, C.-R.G. Preliminary predictive criteria for COVID-19 cytokine storm. Ann. Rheum. Dis. 2020, 80, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e7. [Google Scholar] [CrossRef] [PubMed]
- Mudd, P.A.; Crawford, J.C.; Turner, J.S.; Souquette, A.; Reynolds, D.; Bender, D.; Bosanquet, J.P.; Anand, N.J.; Striker, D.A.; Martin, R.S.; et al. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci. Adv. 2020, 6, eabe3024. [Google Scholar] [CrossRef]
- Kox, M.; Waalders, N.J.B.; Kooistra, E.J.; Gerretsen, J.; Pickkers, P. Cytokine Levels in Critically Ill Patients with COVID-19 and Other Conditions. JAMA 2020, 324, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Ferro, F.; Elefante, E.; Baldini, C.; Bartoloni, E.; Puxeddu, I.; Talarico, R.; Mosca, M.; Bombardieri, S. COVID-19: The new challenge for rheumatologists. Clin. Exp. Rheumatol. 2020, 38, 175–180. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vályi-Nagy, I.; Uher, F.; Rákóczi, É.; Szekanecz, Z. Adaptive Immunity to Viruses: What Did We Learn from SARS-CoV-2 Infection? Int. J. Mol. Sci. 2022, 23, 13951. https://doi.org/10.3390/ijms232213951
Vályi-Nagy I, Uher F, Rákóczi É, Szekanecz Z. Adaptive Immunity to Viruses: What Did We Learn from SARS-CoV-2 Infection? International Journal of Molecular Sciences. 2022; 23(22):13951. https://doi.org/10.3390/ijms232213951
Chicago/Turabian StyleVályi-Nagy, István, Ferenc Uher, Éva Rákóczi, and Zoltán Szekanecz. 2022. "Adaptive Immunity to Viruses: What Did We Learn from SARS-CoV-2 Infection?" International Journal of Molecular Sciences 23, no. 22: 13951. https://doi.org/10.3390/ijms232213951