
High Glucose-Induced Cardiomyocyte Damage Involves Interplay between Endothelin ET-1/ETA/ETB Receptor and mTOR Pathway
 , and
, and
Abstract
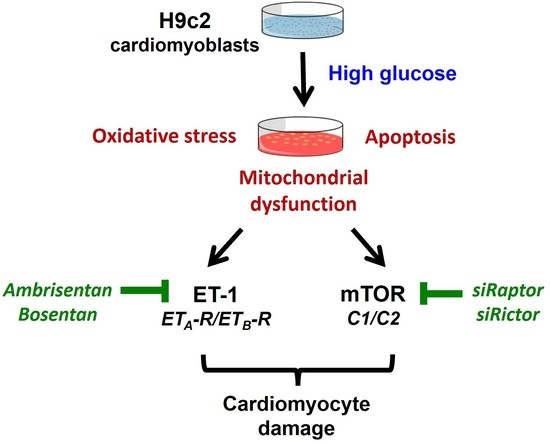
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. HG Treatment Upregulated ET-1/ETA-R/ETB-R/mTOR in H9c2 Cardiomyoblasts
2.2. Treatment with ERAs or Inhibition of mTOR Protected against HG-Induced Oxidative Damage
2.3. Inhibition of mTOR Alleviates HG-Induced Oxidative Stress and Improves the Survival Markers
2.4. Inhibition of mTOR Decreased ET-1 Mediated Mitochondrial Damage in HG-Treated H9c2 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Protein Expression Analysis by Western Blotting
4.5. Measurement of Intracellular ROS Level
4.6. Glucose Uptake Assay
4.7. siRNA Transfection
4.8. Mitochondrial ROS Measurement
4.9. Quantitative Real-Time PCR (qPCR)
4.10. Measurement of Mitochondrial Membrane Potential by JC-1 Staining
4.11. Measurement Labelling
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palazzuoli, A.; Iacoviello, M. Diabetes leading to heart failure and heart failure leading to diabetes: Epidemiological and clinical evidence. Heart Fail Rev. 2022; in press. [Google Scholar] [CrossRef]
- El Hadi, H.; Vettor, R.; Rossato, M. Cardiomyocyte mitochondrial dysfunction in diabetes and its contribution in cardiac arrhythmogenesis. Mitochondrion 2019, 46, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Makrecka-Kuka, M.; Liepinsh, E.; Murray, A.J.; Lemieux, H.; Dambrova, M.; Tepp, K.; Puurand, M.; Kaambre, T.; Han, W.H.; de Goede, P.; et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2020, 228, e13430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Li, W.; Wang, G.; Guo, L.; Jiang, Y.; Kang, Y.J. Hyperglycemia-induced apoptosis in mouse myocardium: Mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 2002, 51, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lin, Q.; Chen, J.; Wei, T.; Li, C.; Zhao, L.; Gao, H.; Zheng, H. High glucose-induced cardiomyocyte death may be linked to unbalanced branched-chain amino acids and energy metabolism. Molecules 2018, 23, 807. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Gintant, G.A.; Miller, R.E.; Davidoff, A.J. High extracellular glucose impairs cardiac E-C coupling in a glycosylation-dependent manner. Am. J. Physiol. 1997, 273, H2876–H2883. [Google Scholar] [CrossRef]
- Sorrentino, A.; Borghetti, G.; Zhou, Y.; Cannata, A.; Meo, M.; Signore, S.; Anversa, P.; Leri, A.; Goichberg, P.; Qanud, K.; et al. Hyperglycemia induces defective Ca2+ homeostasis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H150–H161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Xiang, H.; Zhao, S.; Sang, H.; Lv, F.; Chen, R.; Shu, Z.; Chen, A.F.; Chen, S.; Lu, H. Vildagliptin improves high glucose-induced endothelial mitochondrial dysfunction via inhibiting mitochondrial fission. J. Cell Mol. Med. 2019, 23, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Lin, Y.; Wang, Y.; Wang, Y. High-glucose induces cardiac myocytes apoptosis through Foxo1/GRK2 signaling pathway. Biochem. Biophys. Res. Commun. 2019, 513, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, R.; Kitazumi, K.; Takasaki, C.; Ohnaka, K.; Aimoto, S.; Tasaka, K.; Ohashi, M.; Nawata, H. Presence of non-selective type of endothelin receptor on vascular endothelium and its linkage to vasodilation. FEBS Lett. 1991, 282, 103–106. [Google Scholar] [CrossRef]
- Sakai, S.; Miyauchi, T.; Kobayashi, M.; Yamaguchi, I.; Goto, K.; Sugishita, Y. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature 1996, 384, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Shubeita, H.E.; McDonough, P.M.; Harris, A.N.; Knowlton, K.U.; Glembotski, C.C.; Brown, J.H.; Chien, K.R. Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular myocytes. A paracrine mechanism for myocardial cell hypertrophy. J. Biol. Chem. 1990, 265, 20555–20562. [Google Scholar] [CrossRef]
- Ceylan-Isik, A.F.; Dong, M.; Zhang, Y.; Dong, F.; Turdi, S.; Nair, S.; Yanagisawa, M.; Ren, J. Cardiomyocyte-specific deletion of endothelin receptor A rescues aging-associated cardiac hypertrophy and contractile dysfunction: Role of autophagy. Basic Res. Cardiol. 2013, 108, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Maitland, A.; Weisel, R.D.; Fedak, P.W.; Li, S.H.; Mickle, D.A.; Li, R.K.; Ko, L.; Rao, V. Increased endothelin-1 production in diabetic patients after cardioplegic arrest and reperfusion impairs coronary vascular reactivity: Reversal by means of endothelin antagonism. J. Thorac. Cardiovasc. Surg. 2002, 123, 1114–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Maitland, A.; Weisel, R.D.; Li, S.H.; Fedak, P.W.; Pomroy, N.C.; Mickle, D.A.; Li, R.K.; Ko, L.; Rao, V. Hyperglycemia exaggerates ischemia-reperfusion-induced cardiomyocyte injury: Reversal with endothelin antagonism. J. Thorac. Cardiovasc. Surg. 2002, 123, 1120–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Li, M.; Shi, Z.G.; Feng, Q.Z. Bosentan ameliorates the expression of fibrotic related growth factors and collagen-1 in diabetic mice. Anadolu Kardiyol. Derg. 2012, 12, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zou, R.; Judd, R.L.; Zhong, J. Endothelin-1 receptor blockade prevented the electrophysiological dysfunction in cardiac myocytes of streptozotocin-induced diabetic rats. Endocrine 2006, 30, 121–127. [Google Scholar] [CrossRef]
- Jiang, J.; Yuen, V.; Xiang, H.; McNeill, J.H. Improvement in cardiac function of diabetic rats by bosentan is not associated with changes in the activation of PKC isoforms. Mol. Cell Biochem. 2006, 282, 177–185. [Google Scholar] [CrossRef]
- Cerrato, R.; Cunnington, C.; Crabtree, M.J.; Antoniades, C.; Pernow, J.; Channon, K.M.; Bohm, F. Endothelin-1 increases superoxide production in human coronary artery bypass grafts. Life Sci. 2012, 91, 723–728. [Google Scholar] [CrossRef] [Green Version]
- Briyal, S.; Philip, T.; Gulati, A. Endothelin-A receptor antagonists prevent amyloid-beta-induced increase in ETA receptor expression, oxidative stress, and cognitive impairment. J. Alzheimer’s Dis. 2011, 23, 491–503. [Google Scholar] [CrossRef]
- Ozdemir, R.; Parlakpinar, H.; Polat, A.; Colak, C.; Ermis, N.; Acet, A. Selective endothelin A (ETA) receptor antagonist (BQ-123) reduces both myocardial infarct size and oxidant injury. Toxicology 2006, 219, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Koka, S.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J. Biol. Chem. 2014, 289, 4145–4160. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, H.; Liu, H.; Li, K.; Jia, M.; Su, X. High glucose upregulated vascular smooth muscle endothelin subtype B receptors via inhibition of autophagy in rat superior mesenteric arteries. Ann. Vasc. Surg. 2018, 52, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Gan, L.; Liu, Z.; Liu, L.; Chang, J.R.; Yin, D.C.; Cao, H.L.; Su, X.L.; Smith, W.W. Mitochondrial-derived peptide MOTS-c attenuates vascular calcification and secondary myocardial remodeling via adenosine monophosphate-activated protein kinase signaling pathway. Cardiorenal Med. 2020, 10, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Suhara, T.; Baba, Y.; Shimada, B.K.; Higa, J.K.; Matsui, T. The mTOR signaling pathway in myocardial dysfunction in type 2 diabetes mellitus. Curr. Diabetes Rep. 2017, 17, 38. [Google Scholar] [CrossRef]
- Park, K.R.; Nam, D.; Yun, H.M.; Lee, S.G.; Jang, H.J.; Sethi, G.; Cho, S.K.; Ahn, K.S. Beta-caryophyllene oxide inhibits growth and induces apoptosis through the suppression of PI3K/AKT/mTOR/S6K1 pathways and ROS-mediated MAPKs activation. Cancer Lett. 2011, 312, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Gao, L.; Zucker, I.H. Regulation of Nrf2 signaling pathway in heart failure: Role of extracellular vesicles and non-coding RNAs. Free Radic. Biol. Med. 2021, 167, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wang, F.; Wang, B.; Qiao, Z.; Cui, X.; Zhang, Y.; Jiang, Q.; Liu, M.; Shangguan, J.; Zheng, X.; et al. A novel compound, tanshinol borneol ester, ameliorates pressure overload-induced cardiac hypertrophy by inhibiting oxidative stress via the mTOR/beta-TrCP/NRF2 pathway. Front. Pharmacol. 2022, 13, 830763. [Google Scholar] [CrossRef] [PubMed]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Zhang, Q.; Yu, Y.; Zhang, Y.; Zhang, M.; Liu, Q.; Zhang, E.; Li, S.; Song, G. Oleanolic acid, a novel endothelin A receptor antagonist, alleviated high glucose-induced cardiomyocytes injury. Am. J. Chin. Med. 2018, 46, 1187–1201. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Chen, C.; Gao, J.L.; Liu, M.Y.; Li, S.L.; Xuan, X.C.; Zhang, X.Z.; Zhang, X.Y.; Wei, Y.Y.; Zhen, C.L.; Jin, J.; et al. Mitochondrial fission inhibitors suppress endothelin-1-induced artery constriction. Cell Physiol. Biochem. 2017, 42, 1802–1811. [Google Scholar] [CrossRef]
- Adaniya, S.M.; O-Uchi, J.; Cypress, M.W.; Kusakari, Y.; Jhun, B.S. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am. J. Physiol. Cell Physiol. 2019, 316, C583–C604. [Google Scholar] [CrossRef]
- Jang, W.B.; Park, J.H.; Ji, S.T.; Lee, N.K.; Kim, D.Y.; Kim, Y.J.; Jung, S.Y.; Kang, S.; Lamichane, S.; Lamichane, B.D.; et al. Cytoprotective roles of a novel compound, MHY-1684, against hyperglycemia-induced oxidative stress and mitochondrial dysfunction in human cardiac progenitor cells. Oxidative Med. Cell Longev. 2018, 2018, 4528184. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Maimaitili, Y.; Xie, P.; Wu, J.J.; Wang, J.; Yang, Y.N.; Ma, H.P.; Zheng, H. High glucose concentration abrogates sevoflurane post-conditioning cardioprotection by advancing mitochondrial fission but dynamin-related protein 1 inhibitor restores these effects. Acta Physiol. 2017, 220, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Burkhoff, D.; Weiss, R.G.; Schulman, S.P.; Kalil-Filho, R.; Wannenburg, T.; Gerstenblith, G. Influence of metabolic substrate on rat heart function and metabolism at different coronary flows. Am. J. Physiol. 1991, 261 Pt 2, H741–H750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, D.; Rodrigues, B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am. J. Physiol. Heart. Circ. Physiol. 2006, 291, H1489–H1506. [Google Scholar] [CrossRef] [Green Version]
- Tykocki, N.R.; Watts, S.W. The interdependence of endothelin-1 and calcium: A review. Clin. Sci. 2010, 119, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Lamblin, N.; Fertin, M.; de Groote, P.; Bauters, C. Cardiac remodeling and heart failure after a first anterior myocardial infarction in patients with diabetes mellitus. J. Cardiovasc. Med. 2012, 13, 353–359. [Google Scholar] [CrossRef]
- Ansley, D.M.; Wang, B. Oxidative stress and myocardial injury in the diabetic heart. J. Pathol. 2013, 229, 232–241. [Google Scholar] [CrossRef]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk between oxidative stress and endoplasmic reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: A focus on the protective roles of heme oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Pang, B.; Shi, L.W.; Du, L.J.; Li, Y.C.; Zhang, M.Z.; Ni, Q. Sheng Mai San protects H9C2 cells against hyperglycemia-induced apoptosis. BMC Complement. Altern. Med. 2019, 19, 309. [Google Scholar] [CrossRef]
- Feng, B.; Chen, S.; Chiu, J.; George, B.; Chakrabarti, S. Regulation of cardiomyocyte hypertrophy in diabetes at the transcriptional level. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E1119–E1126. [Google Scholar] [CrossRef] [Green Version]
- Varela, R.; Rauschert, I.; Romanelli, G.; Alberro, A.; Benech, J.C. Hyperglycemia and hyperlipidemia can induce morphophysiological changes in rat cardiac cell line. Biochem. Biophys. Rep. 2021, 26, 100983. [Google Scholar] [CrossRef]
- Studelska, D.R.; Campbell, C.; Pang, S.; Rodnick, K.J.; James, D.E. Developmental expression of insulin-regulatable glucose transporter GLUT-4. Am. J. Physiol. 1992, 263 Pt 1, E102–E106. [Google Scholar] [CrossRef]
- Shao, D.; Tian, R. Glucose transporters in cardiac metabolism and hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [PubMed] [Green Version]
- Abel, E.D. Glucose transport in the heart. Front. Biosci. 2004, 9, 201–215. [Google Scholar] [CrossRef]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Buller, C.L.; Heilig, C.W.; Brosius, F.C., 3rd. GLUT1 enhances mTOR activity independently of TSC2 and AMPK. Am. J. Physiol. Renal Physiol. 2011, 301, F588–F596. [Google Scholar] [CrossRef]
- Buller, C.L.; Loberg, R.D.; Fan, M.H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Guan, K.L.; Brosius, F.C., 3rd. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Cell Physiol. 2008, 295, C836–C843. [Google Scholar] [CrossRef]
- Horinouchi, T.; Terada, K.; Higashi, T.; Miwa, S. Endothelin receptor signaling: New insight into its regulatory mechanisms. J. Pharmacol. Sci. 2013, 123, 85–101. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Evans, T.; Mukherjee, K.; Karmazyn, M.; Chakrabarti, S. Diabetes-induced myocardial structural changes: Role of endothelin-1 and its receptors. J. Mol. Cell Cardiol. 2000, 32, 1621–1629. [Google Scholar] [CrossRef]
- Ott, C.; Jung, T.; Brix, S.; John, C.; Betz, I.R.; Foryst-Ludwig, A.; Deubel, S.; Kuebler, W.M.; Grune, T.; Kintscher, U.; et al. Hypertrophy-reduced autophagy causes cardiac dysfunction by directly impacting cardiomyocyte contractility. Cells 2021, 10, 805. [Google Scholar] [CrossRef]
- Engedal, N.; Torgersen, M.L.; Guldvik, I.J.; Barfeld, S.J.; Bakula, D.; Saetre, F.; Hagen, L.K.; Patterson, J.B.; Proikas-Cezanne, T.; Seglen, P.O.; et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 2013, 9, 1475–1490. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, S.; Chakraborty, T.; Shimizu, S.; Ohta, K.; Nagahama, Y.; Ohta, K. Estrogen and estrogen receptors chauffeur the sex-biased autophagic action in liver. Cell Death Differ. 2020, 27, 3117–3130. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.J.; Borthwick, G.M.; Arthur, H.M. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. Vitr. Cell Dev. Biol. Anim. 2011, 47, 125–131. [Google Scholar] [CrossRef]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Nuamnaichati, N.; Mangmool, S.; Chattipakorn, N.; Parichatikanond, W. Stimulation of GLP-1 receptor inhibits methylglyoxal-induced mitochondrial dysfunctions in H9c2 cardiomyoblasts: Potential role of Epac/PI3K/Akt pathway. Front. Pharmacol. 2020, 11, 805. [Google Scholar] [CrossRef]
- Parichatikanond, W.; Nishimura, A.; Nishida, M.; Mangmool, S. Prolonged stimulation of β2-adrenergic receptor with β2-agonists impairs insulin actions in H9c2 cells. J. Pharmacol. Sci. 2018, 138, 184–191. [Google Scholar] [CrossRef]
- Phosri, S.; Bunrukchai, K.; Parichatikanond, W.; Sato, V.H.; Mangmool, S. Epac is required for exogenous and endogenous stimulation of adenosine A2B receptor for inhibition of angiotensin II-induced collagen synthesis and myofibroblast differentiation. Purinergic Signal. 2018, 14, 141–156. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, S.; Madreiter-Sokolowski, C.T.; Mangmool, S.; Parichatikanond, W. High Glucose-Induced Cardiomyocyte Damage Involves Interplay between Endothelin ET-1/ETA/ETB Receptor and mTOR Pathway. Int. J. Mol. Sci. 2022, 23, 13816. https://doi.org/10.3390/ijms232213816
Pandey S, Madreiter-Sokolowski CT, Mangmool S, Parichatikanond W. High Glucose-Induced Cardiomyocyte Damage Involves Interplay between Endothelin ET-1/ETA/ETB Receptor and mTOR Pathway. International Journal of Molecular Sciences. 2022; 23(22):13816. https://doi.org/10.3390/ijms232213816
Chicago/Turabian StylePandey, Sudhir, Corina T. Madreiter-Sokolowski, Supachoke Mangmool, and Warisara Parichatikanond. 2022. "High Glucose-Induced Cardiomyocyte Damage Involves Interplay between Endothelin ET-1/ETA/ETB Receptor and mTOR Pathway" International Journal of Molecular Sciences 23, no. 22: 13816. https://doi.org/10.3390/ijms232213816