
Sustained Effects of CGRP Blockade on Cortical Spreading Depolarization-Induced Alterations in Facial Heat Pain Threshold, Light Aversiveness, and Locomotive Activity in the Light Environment
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
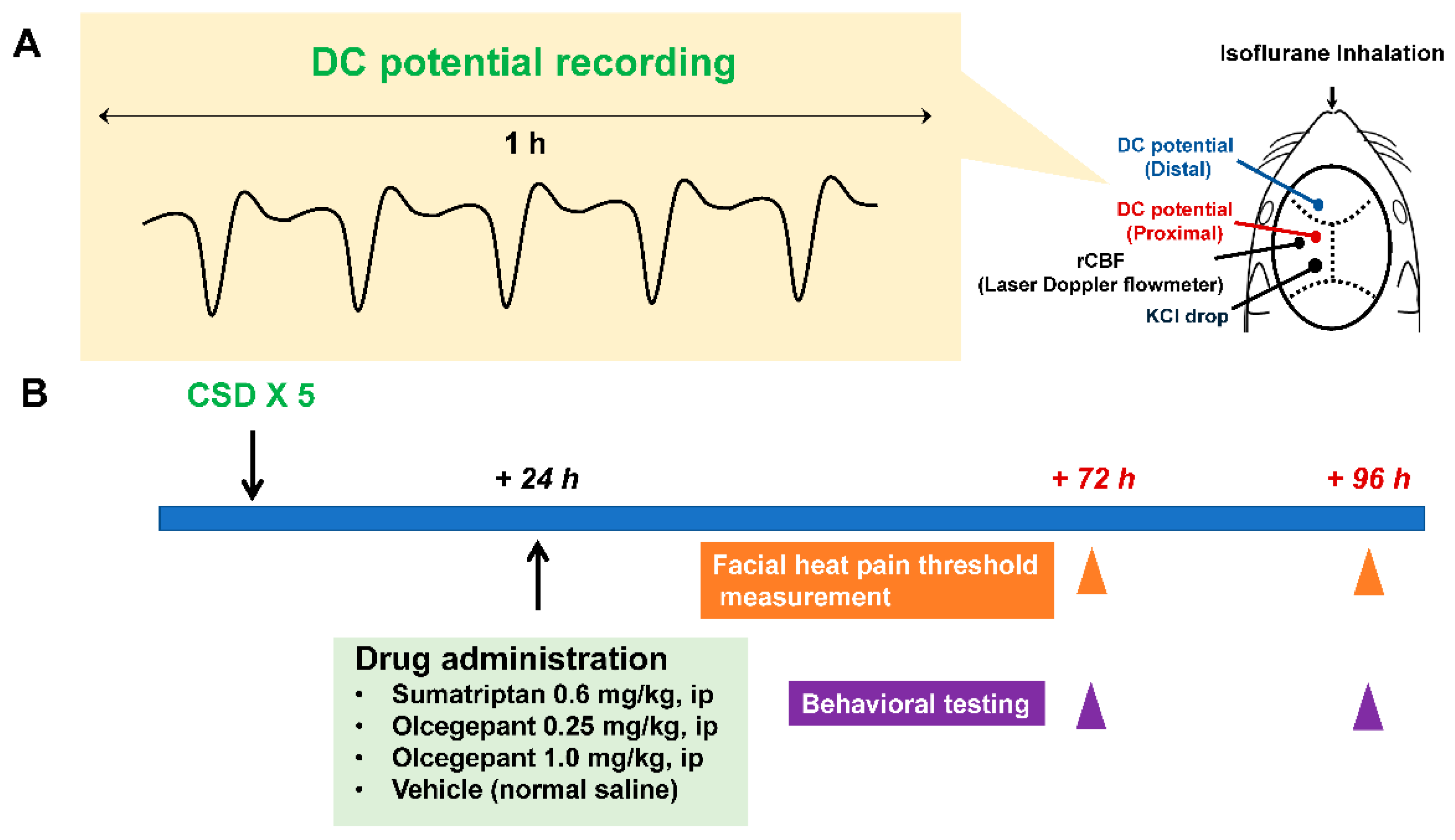
2.1. Experimental Timelines
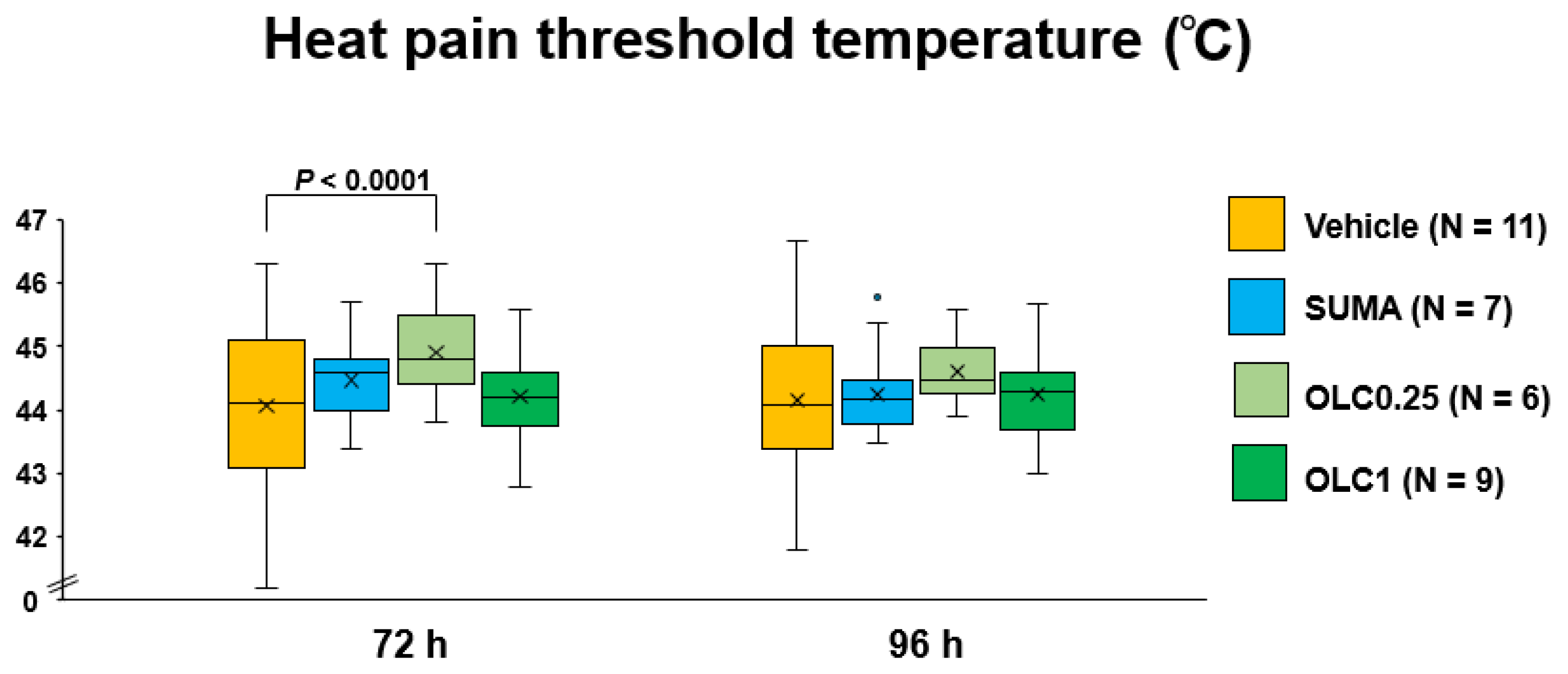
2.2. Temporal Profiles of Heat Pain Threshold Temperature after CSD
2.3. Effects of Sumatriptan and Olcegepant on Total Time Spent in the Light Zone after CSD
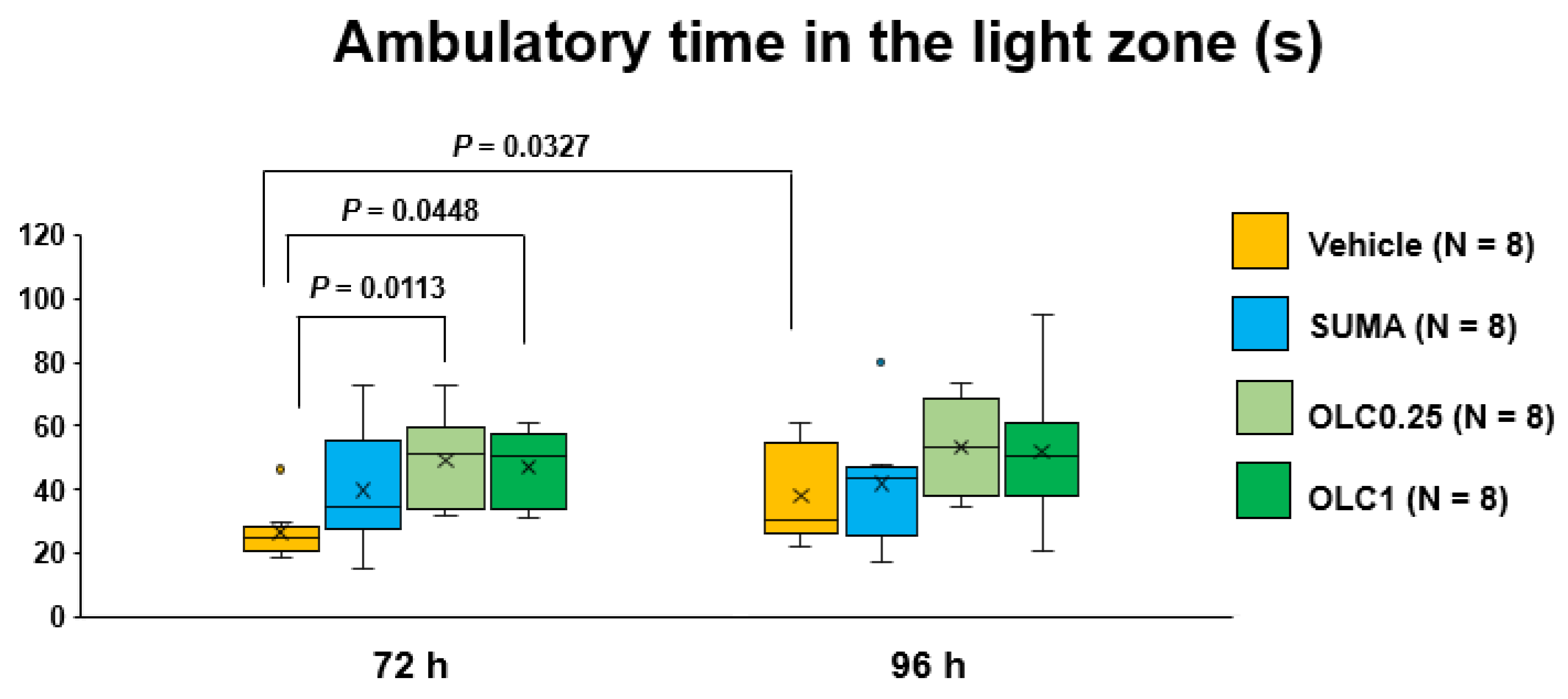
2.4. Effects of Sumatriptan and Olcegepant on Ambulatory Time and Ambulatory Distance in the Light Zone after CSD
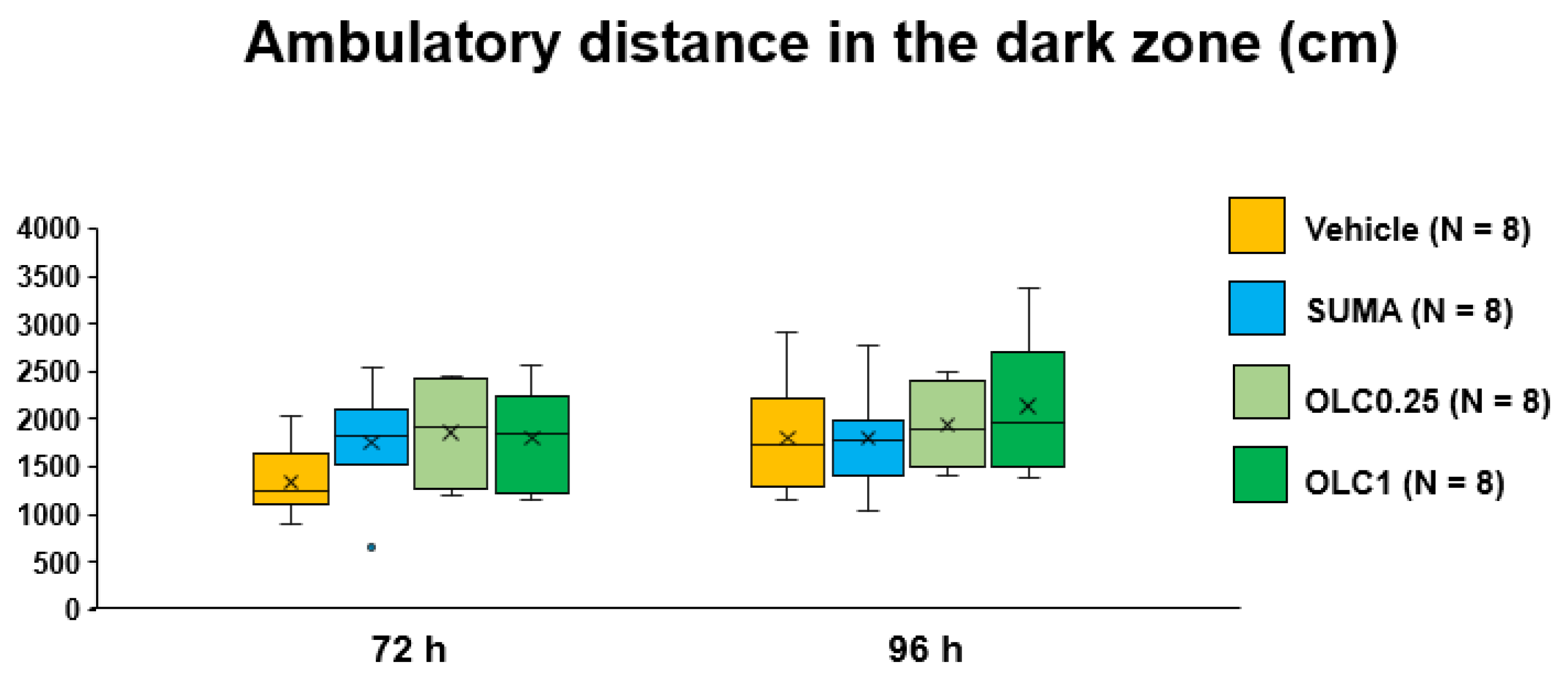
2.5. Effects of Sumatriptan and Olcegepant on Ambulatory Time and Ambulatory Distance in the Dark Zone after CSD
2.6. Comparison of the Average Ambulatory Speed among Experimental Groups
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. CSD Induction
4.3. Drug Administration
4.4. Facial Heat Pain Threshold Temperature Measurement
4.5. Behavioral Analysis in the Light and Dark Zones
4.6. Evaluation of Mouse Locomotion
4.7. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steiner, T.J.; Stovner, L.J.; Jensen, R.; Uluduz, D.; Katsarava, Z.; Lifting The Burden: The Global Campaign against, H. Migraine remains second among the world’s causes of disability, and first among young women: Findings from GBD2019. J. Headache Pain 2020, 21, 137. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M. Migraine. N. Engl. J. Med. 2020, 383, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Lampl, C.; Thomas, H.; Stovner, L.J.; Tassorelli, C.; Katsarava, Z.; Lainez, J.M.; Lanteri-Minet, M.; Rastenyte, D.; Ruiz de la Torre, E.; Andree, C.; et al. Interictal burden attributable to episodic headache: Findings from the Eurolight project. J. Headache Pain 2016, 17, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumori, Y.; Ueda, K.; Komori, M.; Zagar, A.J.; Kim, Y.; Jaffe, D.H.; Takeshima, T.; Hirata, K. Burden of Migraine in Japan: Results of the ObserVational Survey of the Epidemiology, tReatment, and Care Of MigrainE (OVERCOME [Japan]) Study. Neurol. Ther. 2022, 11, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Hubig, L.T.; Smith, T.; Williams, E.; Powell, L.; Johnston, K.; Harris, L.; L’Italien, G.; Coric, V.; Lloyd, A.J.; Lo, S.H. Measuring interictal burden among people affected by migraine: A descriptive survey study. J. Headache Pain 2022, 23, 97. [Google Scholar] [CrossRef]
- Giffin, N.J.; Lipton, R.B.; Silberstein, S.D.; Olesen, J.; Goadsby, P.J. The migraine postdrome: An electronic diary study. Neurology 2016, 87, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Kelman, L. The postdrome of the acute migraine attack. Cephalalgia 2006, 26, 214–220. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Lanteri-Minet, M.; Michel, M.C.; Peres, M.; Shibata, M.; Straube, A.; Wijeratne, T.; Ebel-Bitoun, C.; Constantin, L.; Hitier, S. 21st century headache: Mapping new territory. J. Headache Pain 2021, 22, 19. [Google Scholar] [CrossRef]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; Shuttleworth, C.W.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef]
- Lauritzen, M.; Jorgensen, M.B.; Diemer, N.H.; Gjedde, A.; Hansen, A.J. Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann. Neurol. 1982, 12, 469–474. [Google Scholar] [CrossRef]
- Olesen, J.; Friberg, L.; Olsen, T.S.; Iversen, H.K.; Lassen, N.A.; Andersen, A.R.; Karle, A. Timing and topography of cerebral blood flow, aura, and headache during migraine attacks. Ann. Neurol. 1990, 28, 791–798. [Google Scholar] [CrossRef]
- Olesen, J.; Larsen, B.; Lauritzen, M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann. Neurol. 1981, 9, 344–352. [Google Scholar] [CrossRef]
- Cao, Y.; Welch, K.M.; Aurora, S.; Vikingstad, E.M. Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch. Neurol. 1999, 56, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Andersen, A.R.; Friberg, L.; Olsen, T.S.; Olesen, J. Delayed hyperemia following hypoperfusion in classic migraine. Single photon emission computed tomographic demonstration. Arch. Neurol. 1988, 45, 154–159. [Google Scholar] [CrossRef]
- Bowyer, S.M.; Aurora, K.S.; Moran, J.E.; Tepley, N.; Welch, K.M. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann. Neurol. 2001, 50, 582–587. [Google Scholar] [CrossRef]
- Hadjikhani, N.; Sanchez Del Rio, M.; Wu, O.; Schwartz, D.; Bakker, D.; Fischl, B.; Kwong, K.K.; Cutrer, F.M.; Rosen, B.R.; Tootell, R.B.; et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. USA 2001, 98, 4687–4692. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Levy, D.; Noseda, R.; Kainz, V.; Jakubowski, M.; Burstein, R. Activation of meningeal nociceptors by cortical spreading depression: Implications for migraine with aura. J. Neurosci. 2010, 30, 8807–8814. [Google Scholar] [CrossRef]
- Zhang, X.; Levy, D.; Kainz, V.; Noseda, R.; Jakubowski, M.; Burstein, R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann. Neurol. 2011, 69, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Karatas, H.; Erdener, S.E.; Gursoy-Ozdemir, Y.; Lule, S.; Eren-Kocak, E.; Sen, Z.D.; Dalkara, T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013, 339, 1092–1095. [Google Scholar] [CrossRef]
- Ayata, C. Pearls and pitfalls in experimental models of spreading depression. Cephalalgia 2013, 33, 604–613. [Google Scholar] [CrossRef]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat. Rev. Neurol. 2018, 14, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Mayberg, M.; Langer, R.S.; Zervas, N.T.; Moskowitz, M.A. Perivascular meningeal projections from cat trigeminal ganglia: Possible pathway for vascular headaches in man. Science 1981, 213, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Ashina, M.; Hansen, J.M.; Do, T.P.; Melo-Carrillo, A.; Burstein, R.; Moskowitz, M.A. Migraine and the trigeminovascular system-40 years and counting. Lancet Neurol 2019, 18, 795–804. [Google Scholar] [CrossRef]
- Buzzi, M.G.; Carter, W.B.; Shimizu, T.; Heath, H., 3rd; Moskowitz, M.A. Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology 1991, 30, 1193–1200. [Google Scholar] [CrossRef]
- Messlinger, K.; Hanesch, U.; Kurosawa, M.; Pawlak, M.; Schmidt, R.F. Calcitonin gene related peptide released from dural nerve fibers mediates increase of meningeal blood flow in the rat. Can. J. Physiol. Pharmacol. 1995, 73, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Eltorp, C.T.; Jansen-Olesen, I.; Hansen, A.J. Release of calcitonin gene-related peptide (CGRP) from guinea pig dura mater in vitro is inhibited by sumatriptan but unaffected by nitric oxide. Cephalalgia 2000, 20, 838–844. [Google Scholar] [CrossRef]
- Edvinsson, L.; Goadsby, P.J. Neuropeptides in migraine and cluster headache. Cephalalgia 1994, 14, 320–327. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Edvinsson, L.; Ekman, R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann. Neurol. 1990, 28, 183–187. [Google Scholar] [CrossRef]
- Bullock, C.M.; Wookey, P.; Bennett, A.; Mobasheri, A.; Dickerson, I.; Kelly, S. Peripheral calcitonin gene-related peptide receptor activation and mechanical sensitization of the joint in rat models of osteoarthritis pain. Arthritis Rheumatol. 2014, 66, 2188–2200. [Google Scholar] [CrossRef] [Green Version]
- Chatchaisak, D.; Connor, M.; Srikiatkhachorn, A.; Chetsawang, B. The potentiating effect of calcitonin gene-related peptide on transient receptor potential vanilloid-1 activity and the electrophysiological responses of rat trigeminal neurons to nociceptive stimuli. J. Physiol. Sci. 2018, 68, 261–268. [Google Scholar] [CrossRef]
- Cornelison, L.E.; Hawkins, J.L.; Durham, P.L. Elevated levels of calcitonin gene-related peptide in upper spinal cord promotes sensitization of primary trigeminal nociceptive neurons. Neuroscience 2016, 339, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, S.; Johnson, K.W.; Ossipov, M.H.; Aurora, S.K. CGRP and the Trigeminal System in Migraine. Headache 2019, 59, 659–681. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ren, Y.; Xu, X.; Zou, X.; Fang, L.; Lin, Q. Sensitization of primary afferent nociceptors induced by intradermal capsaicin involves the peripheral release of calcitonin gene-related Peptide driven by dorsal root reflexes. J. Pain 2008, 9, 1155–1168. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hoff, A.O.; Wimalawansa, S.J.; Cote, G.J.; Gagel, R.F.; Westlund, K.N. Arthritic calcitonin/alpha calcitonin gene-related peptide knockout mice have reduced nociceptive hypersensitivity. Pain 2001, 89, 265–273. [Google Scholar] [CrossRef]
- Durham, P.L.; Russo, A.F. Regulation of calcitonin gene-related peptide secretion by a serotonergic antimigraine drug. J. Neurosci. 1999, 19, 3423–3429. [Google Scholar] [CrossRef] [Green Version]
- Limmroth, V.; Katsarava, Z.; Liedert, B.; Guehring, H.; Schmitz, K.; Diener, H.C.; Michel, M.C. An in vivo rat model to study calcitonin gene related peptide release following activation of the trigeminal vascular system. Pain 2001, 92, 101–106. [Google Scholar] [CrossRef]
- Tang, C.; Unekawa, M.; Kitagawa, S.; Takizawa, T.; Kayama, Y.; Nakahara, J.; Shibata, M. Cortical spreading depolarisation-induced facial hyperalgesia, photophobia and hypomotility are ameliorated by sumatriptan and olcegepant. Sci. Rep. 2020, 10, 11408. [Google Scholar] [CrossRef]
- Shibata, M.; Kitagawa, S.; Tang, C.; Unekawa, M.; Kayama, Y.; Shimizu, T.; Nakahara, J.; Suzuki, N. Protracted hypomobility in the absence of trigeminal sensitization after cortical spreading depolarization: Relevance to migraine postdrome. Neurosci. Res. 2021, 172, 80–86. [Google Scholar] [CrossRef]
- Strassman, A.M.; Melo-Carrillo, A.; Houle, T.T.; Adams, A.; Brin, M.F.; Burstein, R. Atogepant - an orally-administered CGRP antagonist-attenuates activation of meningeal nociceptors by CSD. Cephalalgia 2022, 42, 933–943. [Google Scholar] [CrossRef]
- Schwedt, T.J.; Zuniga, L.; Chong, C.D. Low heat pain thresholds in migraineurs between attacks. Cephalalgia 2015, 35, 593–599. [Google Scholar] [CrossRef]
- Stankewitz, A.; Aderjan, D.; Eippert, F.; May, A. Trigeminal nociceptive transmission in migraineurs predicts migraine attacks. J. Neurosci. 2011, 31, 1937–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, L.H.; May, A. The migraine generator revisited: Continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 2016, 139, 1987–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, R.B.; Gandhi, P.; Stokes, J.; Cala, M.L.; Evans, C.J.; Knoble, N.; Gelhorn, H.L.; Revicki, D.; Viswanathan, H.N.; Dodick, D.W. Development and validation of a novel patient-reported outcome measure in people with episodic migraine and chronic migraine: The Activity Impairment in Migraine Diary. Headache 2022, 62, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Ebine, T.; Toriumi, H.; Shimizu, T.; Unekawa, M.; Takizawa, T.; Kayama, Y.; Shibata, M.; Suzuki, N. Alterations in the threshold of the potassium concentration to evoke cortical spreading depression during the natural estrous cycle in mice. Neurosci. Res. 2016, 112, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Vetvik, K.G.; MacGregor, E.A. Sex differences in the epidemiology, clinical features, and pathophysiology of migraine. Lancet Neurol. 2017, 16, 76–87. [Google Scholar] [CrossRef]
- Chen, Y.; Navratilova, E.; Dodick, D.W.; Porreca, F. An Emerging Role for Prolactin in Female-Selective Pain. Trends Neurosci. 2020, 43, 635–648. [Google Scholar] [CrossRef]
- van Casteren, D.S.; Kurth, T.; Danser, A.H.J.; Terwindt, G.M.; MaassenVanDenBrink, A. Sex Differences in Response to Triptans: A Systematic Review and Meta-analysis. Neurology 2021, 96, 162–170. [Google Scholar] [CrossRef]
- Iovino, M.; Feifel, U.; Yong, C.L.; Wolters, J.M.; Wallenstein, G. Safety, tolerability and pharmacokinetics of BIBN 4096 BS, the first selective small molecule calcitonin gene-related peptide receptor antagonist, following single intravenous administration in healthy volunteers. Cephalalgia 2004, 24, 645–656. [Google Scholar] [CrossRef]
- Unekawa, M.; Ikeda, K.; Tomita, Y.; Kawakami, K.; Suzuki, N. Enhanced susceptibility to cortical spreading depression in two types of Na(+),K(+)-ATPase alpha2 subunit-deficient mice as a model of familial hemiplegic migraine 2. Cephalalgia 2018, 38, 1515–1524. [Google Scholar] [CrossRef]
- Kayama, Y.; Shibata, M.; Takizawa, T.; Ibata, K.; Shimizu, T.; Ebine, T.; Toriumi, H.; Yuzaki, M.; Suzuki, N. Functional interactions between transient receptor potential M8 and transient receptor potential V1 in the trigeminal system: Relevance to migraine pathophysiology. Cephalalgia 2018, 38, 833–845. [Google Scholar] [CrossRef]
- Blumstein, L.K.; Crawley, J.N. Further characterization of a simple, automated exploratory model for the anxiolytic effects of benzodiazepines. Pharmacol. Biochem. Behav. 1983, 18, 37–40. [Google Scholar] [CrossRef]
- Onaivi, E.S.; Martin, B.R. Neuropharmacological and physiological validation of a computer-controlled two-compartment black and white box for the assessment of anxiety. Prog. Neuropsychopharmacol. Biol. Psychiatry 1989, 13, 963–976. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitagawa, S.; Tang, C.; Unekawa, M.; Kayama, Y.; Nakahara, J.; Shibata, M. Sustained Effects of CGRP Blockade on Cortical Spreading Depolarization-Induced Alterations in Facial Heat Pain Threshold, Light Aversiveness, and Locomotive Activity in the Light Environment. Int. J. Mol. Sci. 2022, 23, 13807. https://doi.org/10.3390/ijms232213807
Kitagawa S, Tang C, Unekawa M, Kayama Y, Nakahara J, Shibata M. Sustained Effects of CGRP Blockade on Cortical Spreading Depolarization-Induced Alterations in Facial Heat Pain Threshold, Light Aversiveness, and Locomotive Activity in the Light Environment. International Journal of Molecular Sciences. 2022; 23(22):13807. https://doi.org/10.3390/ijms232213807
Chicago/Turabian StyleKitagawa, Satoshi, Chunhua Tang, Miyuki Unekawa, Yohei Kayama, Jin Nakahara, and Mamoru Shibata. 2022. "Sustained Effects of CGRP Blockade on Cortical Spreading Depolarization-Induced Alterations in Facial Heat Pain Threshold, Light Aversiveness, and Locomotive Activity in the Light Environment" International Journal of Molecular Sciences 23, no. 22: 13807. https://doi.org/10.3390/ijms232213807