
Application of Computational Biology and Artificial Intelligence in Drug Design

Abstract
:1. Introduction
2. Computational Biology in Drug Design
2.1. Application of Molecular Mechanics in Drug Design
2.1.1. Application in Investigating the Mechanism of the Target Protein
2.1.2. Application in Molecular Docking
2.1.3. Application in Lead Optimization
2.1.4. Application of Coarse-Grained Models in Drug Design
2.2. Application of QM in Drug Design
3. Computer-Aided Drug Design
3.1. Structure-Based Drug Design
3.1.1. Target Preparation
3.1.2. Binding Site Identification
3.1.3. Compound Library Preparation
3.1.4. Molecular Docking and Scoring
3.1.5. MD Simulations
3.2. Ligand-Based Drug Design
3.2.1. Pharmacophore Modeling
3.2.2. Quantitative Structure–Activity Relationship
4. De Novo Drug Design by Artificial Intelligence
4.1. Overview of the Machine Learning Based de Novo Drug Design
4.2. Overview of de Novo Molecule Generation
4.2.1. Structure-Oriented Generation
4.2.2. Ligand-Oriented Generation
5. Approaches and Techniques in Artificial Intelligence Based de Novo Drug Design
5.1. Datasets in AI-Based de Novo Drug Design
5.2. Descriptors/Feature Representation
5.3. Deep Learning Methods for Molecule Generation
5.4. Machine Learning Methods for Molecular Properties Optimization
5.5. Evaluation
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent advances in computer-aided drug design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyan, K. Pharmaceutical Medicine and Translational Clinical Research. Curr. Sci. 2018, 115, 1403. [Google Scholar]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An Updated Review of Computer-Aided Drug Design and Its Application to COVID-19. BioMed Res. Int. 2021, 2021, 8853056. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Xing, J.; Li, X.; Liu, X.; Fu, Z.; Xiong, Z.; Lu, D.; Wu, X.; Zhao, J.; Tan, X. Artificial intelligence in drug design. Sci. China Life Sci. 2018, 61, 1191–1204. [Google Scholar] [CrossRef]
- Hou, T.; Xu, X. Recent development and application of virtual screening in drug discovery: An overview. Curr. Pharm. Des. 2004, 10, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.G.; Richards, D. Drug Discovery and Development E-Book: Technology in Transition; Elsevier Health Sciences: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Caflisch, A. Molecular dynamics in drug design. Eur. J. Med. Chem. 2015, 91, 4–14. [Google Scholar] [CrossRef]
- Huang, D.; Caflisch, A. The free energy landscape of small molecule unbinding. PLoS Comput. Biol. 2011, 7, e1002002. [Google Scholar] [CrossRef] [Green Version]
- Honarparvar, B.; Govender, T.; Maguire, G.E.; Soliman, M.E.; Kruger, H.G. Integrated approach to structure-based enzymatic drug design: Molecular modeling, spectroscopy, and experimental bioactivity. Chem. Rev. 2014, 114, 493–537. [Google Scholar] [CrossRef] [PubMed]
- Labanowski, J.K.; Andzelm, J.W. Density Functional Methods in Chemistry; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef] [PubMed]
- Chivian, D.; Robertson, T.; Bonneau, R.; Baker, D. Ab initio methods. Meth. Biochem. Anal. 2003, 44, 547–558. [Google Scholar] [CrossRef]
- Sebastiani, D.; Rothlisberger, U. Advances in Density-Functional-Based Modeling Techniques-Recent Extensions of the Car-Parrinello Approach. Meth. Princ. Med. Chem. 2003, 17, 5–40. [Google Scholar] [CrossRef]
- Veselovsky, A.; Ivanov, A. Strategy of computer-aided drug design. Curr. Drug Targets-Infect. Disord. 2003, 3, 33–40. [Google Scholar] [CrossRef]
- Surabhi, S.; Singh, B. Computer aided drug design: An overview. J. Drug Deliv. Ther. 2018, 8, 504–509. [Google Scholar] [CrossRef] [Green Version]
- Kore, P.P.; Mutha, M.M.; Antre, R.V.; Oswal, R.J.; Kshirsagar, S.S. Computer-aided drug design: An innovative tool for modeling. Open J. Med. Chem. 2012, 2, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provazník, I.; Choi, I. Computer aided drug design: Success and limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef]
- Yu, W.; MacKerell, A.D. Computer-aided drug design methods. In Antibiotics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 85–106. [Google Scholar]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Banegas-Luna, A.-J.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H. A review of ligand-based virtual screening web tools and screening algorithms in large molecular databases in the age of big data. Future Med. Chem. 2018, 10, 2641–2658. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Sohraby, F.; Bagheri, M.; Aryapour, H. Performing an in silico repurposing of existing drugs by combining virtual screening and molecular dynamics simulation. In Computational Methods for Drug Repurposing; Springer: Berlin/Heidelberg, Germany, 2019; pp. 23–43. [Google Scholar]
- Issa, N.T.; Kruger, J.; Byers, S.W.; Dakshanamurthy, S. Drug repurposing a reality: From computers to the clinic. Expert Rev. Clin. Pharmacol. 2013, 6, 95–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duch, W.; Swaminathan, K.; Meller, J. Artificial intelligence approaches for rational drug design and discovery. Curr. Pharm. Des. 2007, 13, 1497–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertrudes, J.C.; Maltarollo, V.G.; Silva, R.; Oliveira, P.R.; Honorio, K.M.; Da Silva, A. Machine learning techniques and drug design. Curr. Med. Chem. 2012, 19, 4289–4297. [Google Scholar] [CrossRef]
- Lima, A.N.; Philot, E.A.; Trossini, G.H.G.; Scott, L.P.B.; Maltarollo, V.G.; Honorio, K.M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 2016, 11, 225–239. [Google Scholar] [CrossRef]
- Klambauer, G.; Hochreiter, S.; Rarey, M. Machine Learning in Drug Discovery. J. Chem. Inf. Model. 2019, 59, 945–946. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Tan, J.; Han, D.; Zhu, H. From machine learning to deep learning: Progress in machine intelligence for rational drug discovery. Drug Discov. Today 2017, 22, 1680–1685. [Google Scholar] [CrossRef]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep learning in drug discovery. Mol. Inform. 2016, 35, 3–14. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Carpenter, K.A.; Huang, X. Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: A review. Curr. Pharm. Des. 2018, 24, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Mendolia, I.; Contino, S.; Perricone, U.; Ardizzone, E.; Pirrone, R. Convolutional architectures for virtual screening. BMC Bioinform. 2020, 21, 310. [Google Scholar] [CrossRef] [PubMed]
- Bohr, H. Drug discovery and molecular modeling using artificial intelligence. In Artificial Intelligence in Healthcare; Elsevier: Amsterdam, The Netherlands, 2020; pp. 61–83. [Google Scholar]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Yu, L.; Zhang, N.; Peng, C.; Su, G.; Jing, Y.; Zhang, L.; Wu, T.; Cheng, J.; Guo, Q. The advanced development of molecular targeted therapy for hepatocellular carcinoma. Cancer Biol. Med. 2022, 19, 802–817. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, W.; Nielsen, S.V.; Lindorff-Larsen, K.; Hartmann-Petersen, R.; Ellgaard, L. Bioinformatics analysis identifies several intrinsically disordered human E3 ubiquitin-protein ligases. PeerJ 2016, 4, e1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X. Bioinformatics and drug discovery. Curr. Top. Med. Chem. 2017, 17, 1709–1726. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Luo, H.; Huang, Y.; Fang, C.; Zhao, L.; Li, P.; Zhong, C.; Liu, F. AURKB, CHEK1 and NEK2 as the Potential Target Proteins of Scutellaria barbata on Hepatocellular Carcinoma: An Integrated Bioinformatics Analysis. Int. J. Gen. Med. 2021, 14, 3295. [Google Scholar] [CrossRef]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Ma, N.; Tian, J.; Shao, Y.; Zhu, B.; Wong, Y.K.; Liang, Z.; Zou, C.; Wang, J. Target identification of natural medicine with chemical proteomics approach: Probe synthesis, target fishing and protein identification. Signal Transduct. Target. Ther. 2020, 5, 72. [Google Scholar] [CrossRef]
- Guo, J.; Ning, L.; Ren, H.; Liu, H.; Yao, X. Influence of the pathogenic mutations T188K/R/A on the structural stability and misfolding of human prion protein: Insight from molecular dynamics simulations. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- Hasan, A.S.; Mohammed, F.Q.; Takz, M.M. Design and synthesis of graphene oxide-based glass substrate and its antimicrobial activity against MDR Bacterial Pathogens. J. Mech. Eng. Res. Dev. 2020, 43, 11–17. [Google Scholar]
- Barone, V.; Improta, R.; Rega, N. Computation of protein pK’s values by an integrated density functional theory/polarizable continuum model approach. Theor. Chem. Acc. 2004, 111, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Souza, P.C.; Thallmair, S.; Marrink, S.J.; Mera-Adasme, R. An allosteric pathway in copper, zinc superoxide dismutase unravels the molecular mechanism of the G93A amyotrophic lateral sclerosis-linked mutation. J. Phys. Chem. Lett. 2019, 10, 7740–7744. [Google Scholar] [CrossRef] [PubMed]
- Moreira, C.; Ramos, M.J.; Fernandes, P.A. Reaction mechanism of Mycobacterium Tuberculosis glutamine synthetase using quantum mechanics/molecular mechanics calculations. Chem. A Eur. J. 2016, 22, 9218–9225. [Google Scholar] [CrossRef]
- Peters, M.B.; Raha, K.; Merz, K. Quantum mechanics in structure-based drug design. Curr. Opin. Drug Discov. Dev. 2006, 9, 370. [Google Scholar]
- Barman, T.K.; Hazarika, A.K.; Kalita, U.; Dhar, R.; Borah, L.; Chetri, S.; Ghosh, S.S. Epidemiology of Anti-HIV Drug Resistance: Quantum Mechanics (Qm) and Molecular Mechanics (MM) Studies into the Binding of 6-Aminoquinoline Molecules within HIV Protein (1AJX) and Its Economic Implication; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Xu, D.; Zhou, Y.; Xie, D.; Guo, H. Antibiotic Binding to Monozinc CphA β-Lactamase from Aeromonas hydropila: Quantum Mechanical/Molecular Mechanical and Density Functional Theory Studies. J. Med. Chem. 2005, 48, 6679–6689. [Google Scholar] [CrossRef]
- Hassan, A.U.; Sumrra, S.H. Exploring the bioactive sites of new sulfonamide metal chelates for multi-drug resistance: An experimental versus theoretical design. J. Inorg. Organomet. Polym. Mater. 2022, 32, 513–535. [Google Scholar] [CrossRef]
- Ode, H.; Matsuyama, S.; Hata, M.; Hoshino, T.; Kakizawa, J.; Sugiura, W. Mechanism of drug resistance due to N88S in CRF01_AE HIV-1 protease, analyzed by molecular dynamics simulations. J. Med. Chem. 2007, 50, 1768–1777. [Google Scholar] [CrossRef] [Green Version]
- Amusengeri, A.; Tata, R.B.; Bishop, Ö.T. Understanding the pyrimethamine drug resistance mechanism via combined molecular dynamics and dynamic residue network analysis. Molecules 2020, 25, 904. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Guvench, O. Molecular mechanics. Curr. Pharm. Des. 2014, 20, 3281–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekono, B.D.; Sona, A.N.; Eni, D.B.; Owono, L.C.; Megnassan, E.; Ntie-Kang, F. Molecular mechanics approaches for rational drug design: Forcefields and solvation models. Phys. Sci. Rev. 2021, 20190128. [Google Scholar]
- Williams-Noonan, B.J.; Yuriev, E.; Chalmers, D.K. Free Energy Methods in Drug Design: Prospects of “Alchemical Perturbation” in Medicinal Chemistry. J. Med. Chem. 2018, 61, 638–649. [Google Scholar] [CrossRef]
- Allen, M.P. Introduction to molecular dynamics simulation. Comput. Soft Matter: Synth. Polym. Proteins 2004, 23, 1–28. [Google Scholar]
- Moroy, G.; Sperandio, O.; Rielland, S.; Khemka, S.; Druart, K.; Goyal, D.; Perahia, D.; Miteva, M.A. Sampling of conformational ensemble for virtual screening using molecular dynamics simulations and normal mode analysis. Future Med. Chem. 2015, 7, 2317–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Whittig, L.; Allardice, W. X-ray diffraction techniques. Meth. Soil Anal. Part 1 Phys. Mineral. Meth. 1986, 5, 331–362. [Google Scholar] [CrossRef]
- Javier, G.N.; Christopher, G.T. Cryo-Electron Microscopy: Moving Beyond X-Ray Crystal Structures for Drug Receptors and Drug Development. Ann. Rev. Pharmacol. Toxicol. 2020, 60, 51–71. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.-C.; McMullan, G.; Scheres, S.H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef]
- Weissenberger, G.; Henderikx, R.J.M.; Peters, P.J. Understanding the invisible hands of sample preparation for cryo-EM. Nat. Methods 2021, 18, 463–471. [Google Scholar] [CrossRef]
- Krieger, E.; Nabuurs, S.B.; Vriend, G. Homology modeling. Meth. Biochem. Anal. 2003, 44, 509–524. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- AlQuraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr. Atomistic models and force fields. In Computational Biochemistry and Biophysics; CRC Press: Boca Raton, FL, USA, 2001; pp. 19–50. [Google Scholar]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [Green Version]
- Senftle, T.P.; Hong, S.; Islam, M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M. The ReaxFF reactive force-field: Development, applications and future directions. NPJ Comput. Mater. 2016, 2, 15011. [Google Scholar] [CrossRef] [Green Version]
- Dauber-Osguthorpe, P.; Hagler, A.T. Biomolecular force fields: Where have we been, where are we now, where do we need to go and how do we get there? J. Comput. Aided Mol. Des. 2019, 33, 133–203. [Google Scholar] [CrossRef]
- Lee, M.; Kolev, V.; Warshel, A. Validating a Coarse-Grained Voltage Activation Model by Comparing Its Performance to the Results of Monte Carlo Simulations. J. Phys. Chem. B 2017, 121, 11284–11291. [Google Scholar] [CrossRef]
- Vorobyov, I.; Kim, I.; Chu, Z.T.; Warshel, A. Refining the treatment of membrane proteins by coarse-grained models. Proteins: Struct. Funct. Bioinform. 2016, 84, 92–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikoshi, N.; Hwang, S.; Gati, C.; Matsui, T.; Castillo-Orellana, C.; Raub, A.G.; Garcia, A.A.; Jabbarpour, F.; Batyuk, A.; Broweleit, J. Long-range structural defects by pathogenic mutations in most severe glucose-6-phosphate dehydrogenase deficiency. Proc. Natl. Acad. Sci. USA 2021, 118, e2022790118. [Google Scholar] [CrossRef] [PubMed]
- Budaitis, B.G.; Jariwala, S.; Rao, L.; Yue, Y.; Sept, D.; Verhey, K.J.; Gennerich, A. Pathogenic mutations in the kinesin-3 motor KIF1A diminish force generation and movement through allosteric mechanisms. J. Cell Biol. 2021, 220, e202004227. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Korovesis, D.; Needham, S.R.; Tynan, C.J.; Sagawa, S.; Roberts, S.K.; Kuzmanic, A.; Ortiz-Zapater, E.; Jain, P.; Roovers, R.C. The architecture of EGFR’s basal complexes reveals autoinhibition mechanisms in dimers and oligomers. Nat. Commun. 2018, 9, 4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Wang, D.D.; Yan, H. Genotype-determined EGFR-RTK heterodimerization and its effects on drug resistance in lung Cancer treatment revealed by molecular dynamics simulations. BMC Mol. Cell Biol. 2021, 22, 34. [Google Scholar] [CrossRef] [PubMed]
- Rahnasto-Rilla, M.; Tyni, J.; Huovinen, M.; Jarho, E.; Kulikowicz, T.; Ravichandran, S.; Bohr, V.A.; Ferrucci, L.; Lahtela-Kakkonen, M.; Moaddel, R. Natural polyphenols as sirtuin 6 modulators. Sci. Rep. 2018, 8, 4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Han, M.; Tran, K.; Patil, N.A.; Ma, W.; Roberts, K.D.; Xiao, M.; Sommer, B.; Schreiber, F.; Wang, L. An Intelligent Strategy with All-Atom Molecular Dynamics Simulations for the Design of Lipopeptides against Multidrug-Resistant Pseudomonas aeruginosa. J. Med. Chem. 2022, 65, 10001–10013. [Google Scholar] [CrossRef]
- Cavasotto, C.; Orry, A.J.W. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.J.; Shoichet, B.K. ZINC− a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polgár, T.; Keseru, G.M. Integration of virtual and high throughput screening in lead discovery settings. Comb. Chem. High Throughput Screen. 2011, 14, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Vujanac, M.; Southall, N.; Stebbins, C.E. Inhibitors of the Yersinia protein tyrosine phosphatase through high throughput and virtual screening approaches. Bioorganic Med. Chem. Lett. 2013, 23, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Mittal, A.; Patel, K.; Gatuz, J.L.; Truong, L.; Torres, J.; Mulhearn, D.C.; Johnson, M.E. Identification of novel drug scaffolds for inhibition of SARS-CoV 3-Chymotrypsin-like protease using virtual and high-throughput screenings. Bioorganic Med. Chem. 2014, 22, 167–177. [Google Scholar] [CrossRef]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.; Jewsbury, P.; Essex, J. A review of protein-small molecule docking methods. J. Comput. Aided Mol. Des. 2002, 16, 151–166. [Google Scholar] [CrossRef]
- Schulz-Gasch, T.; Stahl, M. Binding site characteristics in structure-based virtual screening: Evaluation of current docking tools. J. Mol. Model. 2003, 9, 47–57. [Google Scholar] [CrossRef]
- Cross, J.B.; Thompson, D.C.; Rai, B.K.; Baber, J.C.; Fan, K.Y.; Hu, Y.; Humblet, C. Comparison of several molecular docking programs: Pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2009, 49, 1455–1474. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kancharla, S.; Jena, M.K. In silico virtual screening-based study of nutraceuticals predicts the therapeutic potentials of folic acid and its derivatives against COVID-19. VirusDisease 2021, 32, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T.; Poso, A. Binding affinity via docking: Fact and fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste makes waste: A critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res. Rev. 2022, 42, 744–769. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Podobas, A.; Gadioli, D.; Vitali, E.; Palermo, G.; Markidis, S. A review on parallel virtual screening softwares for high-performance computers. Pharmaceuticals 2022, 15, 63. [Google Scholar] [CrossRef]
- Rastelli, G.; Pinzi, L. Refinement and Rescoring of Virtual Screening Results. Front. Chem. 2019, 7, 498. [Google Scholar] [CrossRef] [Green Version]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Schmidt, B.; Voss, G.; Müller-Wittig, W. Molecular dynamics simulations on commodity GPUs with CUDA. In Proceedings of the International Conference on High-Performance Computing, Goa, India, 18–21 December 2007; pp. 185–196. [Google Scholar]
- Ramalingam, G.; Reps, T. On the computational complexity of dynamic graph problems. Theor. Comput. Sci. 1996, 158, 233–277. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Ahmad, I.; Jadhav, H.; Shinde, Y.; Jagtap, V.; Girase, R.; Patel, H. Optimizing Bedaquiline for cardiotoxicity by structure based virtual screening, DFT analysis and molecular dynamic simulation studies to identify selective MDR-TB inhibitors. Silico Pharmacol. 2021, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Chanaga, E.E.; Betancourt-Conde, I.; Hernández-Campos, A.; Téllez-Valencia, A.; Castillo, R. In silico hit optimization toward AKT inhibition: Fragment-based approach, molecular docking and molecular dynamics study. J. Biomol. Struct. Dyn. 2019, 37, 4301–4311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Feng, L.-J.; Huang, Y.; Wu, D.; Li, Z.; Zhou, Q.; Wu, Y.; Luo, H.B. Discovery of Novel Phosphodiesterase-2A Inhibitors by Structure-Based Virtual Screening, Structural Optimization, and Bioassay. J. Chem. Inf. Model. 2017, 57, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Ruskamo, S.; Yadav, R.P.; Sharma, S.; Lehtimäki, M.; Laulumaa, S.; Aggarwal, S.; Simons, M.; Bürck, J.; Ulrich, A.S.; Juffer, A.H. Atomic resolution view into the structure–function relationships of the human myelin peripheral membrane protein P2. Acta Crystallogr. Sect. D: Biol.Crystallogr. 2014, 70, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Singh, N.; Li, W. Recent advances in coarse-grained models for biomolecules and their applications. Int. J. Mol. Sci. 2019, 20, 3774. [Google Scholar] [CrossRef] [Green Version]
- Vicatos, S.; Rychkova, A.; Mukherjee, S.; Warshel, A. An effective Coarse-grained model for biological simulations: Recent refinements and validations. Proteins: Struct. Funct. Bioinform. 2014, 82, 1168–1185. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Chen, G.; Zhang, H.; An, K.; Xu, P.; Du, Y.; Ye, R.D.; Saha, A.; Zhang, A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Mondal, D.; Du, Y.; Ye, R.D.; Warshel, A. Exploring the Activation Process of the β2AR-Gs Complex. J. Am. Chem. Soc. 2021, 143, 11044–11051. [Google Scholar] [CrossRef]
- Mukherjee, S.; Warshel, A. Electrostatic origin of the mechanochemical rotary mechanism and the catalytic dwell of F1-ATPase. Proc. Natl. Acad. Sci. USA 2011, 108, 20550–20555. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.; Warshel, A. Revisiting the protomotive vectorial motion of F0-ATPase. Proc. Natl. Acad. Sci. USA 2019, 116, 19484–19489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Thirumalai, D. Dissecting the kinematics of the kinesin step. Structure 2012, 20, 628–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warshel, A.; Sharma, P.K.; Kato, M.; Parson, W.W. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta Proteins Proteom. 2006, 1764, 1647–1676. [Google Scholar] [CrossRef] [PubMed]
- Marrone, T.J.; Briggs, A.J.M.; McCammon, J.A. Structure-based drug design: Computational advances. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 71–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raha, K.; Peters, M.B.; Wang, B.; Yu, N.; Wollacott, A.M.; Westerhoff, L.M.; Merz, K.M. The role of quantum mechanics in structure-based drug design. Drug Discov. Today 2007, 12, 725–731. [Google Scholar] [CrossRef]
- Enyedy, I.J.; Egan, W.J. Can we use docking and scoring for hit-to-lead optimization? J. Comput. Aided Mol. Des. 2008, 22, 161–168. [Google Scholar] [CrossRef]
- Guterres, H.; Im, W. Improving protein-ligand docking results with high-throughput molecular dynamics simulations. J. Chem. Inf. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Berishvili, V.; Kuimov, A.; Voronkov, A.; Radchenko, E.; Kumar, P.; Choonara, Y.; Pillay, V.; Kamal, A.; Palyulin, V. Discovery of novel tankyrase inhibitors through molecular docking-based virtual screening and molecular dynamics simulation studies. Molecules 2020, 25, 3171. [Google Scholar] [CrossRef]
- Halgren, T.A.; Damm, W. Polarizable force fields. Curr. Opin. Struct. Biol. 2001, 11, 236–242. [Google Scholar] [CrossRef]
- Arodola, O.A.; Soliman, M.E. Quantum mechanics implementation in drug-design workflows: Does it really help? Drug Des. Dev. Ther. 2017, 11, 2551. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.J.; Santos-Martins, D.; Russo, N.; Ramos, M.J.; Fernandes, P.A. Enzymatic flexibility and reaction rate: A QM/MM study of HIV-1 protease. ACS Catal. 2015, 5, 5617–5626. [Google Scholar] [CrossRef]
- Uddin, N.; Ahmed, S.; Khan, A.; Hoque, M.M.; Halim, M.A. Halogenated derivatives of methotrexate as human dihydrofolate reductase inhibitors in cancer chemotherapy. J. Biomol. Struct. Dyn. 2019, 38, 901–917. [Google Scholar] [CrossRef] [PubMed]
- Nakliang, P.; Lazim, R.; Chang, H.; Choi, S. Multiscale molecular modeling in G protein-coupled receptor (GPCR)-ligand studies. Biomolecules 2020, 10, 631. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, S.; Zhang, Y. Catalytic reaction mechanism of acetylcholinesterase determined by Born-Oppenheimer ab initio QM/MM molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 8817–8825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Fang, L.; Liu, J.; Zhan, C.-G. Reaction pathway and free energy profile for butyrylcholinesterase-catalyzed hydrolysis of acetylcholine. J. Phys. Chem. B 2011, 115, 1315–1322. [Google Scholar] [CrossRef]
- Cheng, Y.; Cheng, X.; Radić, Z.; McCammon, J.A. Acetylcholinesterase: Mechanisms of covalent inhibition of wild-type and H447I mutant determined by computational analyses. J. Am. Chem. Soc. 2007, 129, 6562–6570. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Y.; Zhan, C.-G. Reaction pathway and free-energy barrier for reactivation of dimethylphosphoryl-inhibited human acetylcholinesterase. J. Phys. Chem. B 2009, 113, 16226–16236. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, L.; Gardebien, F. Structure-affinity properties of a high-affinity ligand of FKBP12 studied by molecular simulations of a binding intermediate. PLoS ONE 2014, 9, e114610. [Google Scholar] [CrossRef]
- Tosso, R.D.; Andujar, S.A.; Gutierrez, L.; Angelina, E.; Rodriguez, R.; Nogueras, M.; Baldoni, H.; Suvire, F.D.; Cobo, J.; Enriz, R.D. Molecular modeling study of dihydrofolate reductase inhibitors. Molecular dynamics simulations, quantum mechanical calculations, and experimental corroboration. J. Chem. Inf. Model. 2013, 53, 2018–2032. [Google Scholar] [CrossRef]
- Cho, A.E.; Guallar, V.; Berne, B.J.; Friesner, R. Importance of accurate charges in molecular docking: Quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005, 26, 915–931. [Google Scholar] [CrossRef] [Green Version]
- Raha, K.; Merz, K.M. Large-scale validation of a quantum mechanics based scoring function: Predicting the binding affinity and the binding mode of a diverse set of protein-ligand complexes. J. Med. Chem. 2005, 48, 4558–4575. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Gleeson, D. QM/MM calculations in drug discovery: A useful method for studying binding phenomena? J. Chem. Inf. Model. 2009, 49, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, Y.; Lu, G. Recent development in quantum mechanics/molecular mechanics modeling for materials. Int. J. Multiscale Comput. Eng. 2012, 10, 65–82. [Google Scholar] [CrossRef] [Green Version]
- Cavasotto, C.N.; Adler, N.S.; Aucar, M.G. Quantum chemical approaches in structure-based virtual screening and lead optimization. Front. Chem. 2018, 6, 188. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.; McNamara, J.P.; Hillier, I.H.; Bryce, R.A. Assessment of QM/MM scoring functions for molecular docking to HIV-1 protease. J. Chem. Inf. Model. 2009, 49, 913–924. [Google Scholar] [CrossRef]
- Kim, M.; Cho, A.E. Incorporating QM and solvation into docking for applications to GPCR targets. Phys. Chem. Chem. Phys. 2016, 18, 28281–28289. [Google Scholar] [CrossRef] [PubMed]
- Chaskar, P.; Zoete, V.; Röhrig, U.F. On-the-Fly QM/MM Docking with Attracting Cavities. J. Chem. Inf. Model. 2017, 57, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.D.; Love, P.J.; Aspuru-Guzik, A. Computational complexity in electronic structure. Phys. Chem. Chem. Phys. 2013, 15, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Orús, R.; Latorre, J.I. Universality of entanglement and quantum-computation complexity. Phys. Rev. A 2004, 69, 052308. [Google Scholar] [CrossRef] [Green Version]
- Senn, H.M.; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef]
- Wilson, E.; Vant, J.; Layton, J.; Boyd, R.; Lee, H.; Turilli, M.; Hernández, B.; Wilkinson, S.; Jha, S.; Gupta, C. Large-Scale Molecular Dynamics Simulations of Cellular Compartments. In Structure and Function of Membrane Proteins; Springer: Berlin/Heidelberg, Germany, 2021; pp. 335–356. [Google Scholar]
- Hoque, I.; Chatterjee, A.; Bhattacharya, S.; Biswas, R. An approach of computer-aided drug design (CADD) tools for in silico pharmaceutical drug design and development. Int. J. Adv. Res. Biol. Sci. 2017, 4, 60–71. [Google Scholar] [CrossRef]
- McKenna, F.; Martin, M.; Bird, H.; Wright, V. Captopril. Br. Med. J. 1983, 287, 1299. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Nascimento, I.J.; De Aquino, T.M.; Da Silva-Júnior, E.F. Drug repurposing: A strategy for discovering inhibitors against emerging viral infections. Curr. Med. Chem. 2021, 28, 2887–2942. [Google Scholar] [CrossRef]
- Huang, H.-J.; Yu, H.W.; Chen, C.-Y.; Hsu, C.-H.; Chen, H.-Y.; Lee, K.-J.; Tsai, F.-J.; Chen, C.Y.-C. Current developments of computer-aided drug design. J. Taiwan Instit. Chem. Eng. 2010, 41, 623–635. [Google Scholar] [CrossRef]
- Druker, B.J.; Lydon, N.B. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin. Investig. 2000, 105, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Van Drie, J.H. Computer-aided drug design: The next 20 years. J. Comput. Aided Mol. Des. 2007, 21, 591–601. [Google Scholar] [CrossRef]
- Athanasiou, C.; Cournia, Z. From computers to bedside: Computational chemistry contributing to FDA approval. In Biomolecular Simulations in Structure-Based Drug Discover; Gervasio, F.L., Spiwok, V., Eds.; WILEY-VCH: Weinheim, Germany, 2018; Volume 75, pp. 163–203. [Google Scholar]
- Macalino, S.J.Y.; Basith, S.; Clavio, N.A.B.; Chang, H.; Kang, S.; Choi, S. Evolution of in silico strategies for protein-protein interaction drug discovery. Molecules 2018, 23, 1963. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-based drug design strategies and challenges. Curr. Top. Med. Chem. 2018, 18, 998–1006. [Google Scholar] [CrossRef]
- Lerner, E.; Barth, A.; Hendrix, J.; Ambrose, B.; Birkedal, V.; Blanchard, S.C.; Börner, R.; Chung, H.S.; Cordes, T.; Craggs, T.D. FRET-based dynamic structural biology: Challenges, perspectives and an appeal for open-science practices. Elife 2021, 10, e60416. [Google Scholar] [CrossRef]
- Lee, J.; Freddolino, P.L.; Zhang, Y. Ab initio protein structure prediction. In From Protein Structure to Function with Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 3–35. [Google Scholar]
- Pan, L.; Gardner, C.L.; Pagliai, F.A.; Gonzalez, C.F.; Lorca, G.L. Identification of the tolfenamic acid binding pocket in PrbP from Liberibacter asiaticus. Front. Microbiol. 2017, 8, 1591. [Google Scholar] [CrossRef] [Green Version]
- Hetényi, C.; van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, N.M.; Alhossary, A.A.; Mu, Y.; Kwoh, C.-K. Protein-ligand blind docking using QuickVina-W with inter-process spatio-temporal integration. Sci. Rep. 2017, 7, 15451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Grimm, M.; Dai, W.-t.; Hou, M.-c.; Xiao, Z.-X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002, 11, 1729–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorga, B.; Herlem, D.; Barré, E.; Guillou, C. Acetylcholine nicotinic receptors: Finding the putative binding site of allosteric modulators using the “blind docking” approach. J. Mol. Model. 2006, 12, 366–372. [Google Scholar] [CrossRef]
- Liang, J.; Karagiannis, C.; Pitsillou, E.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.C. Site mapping and small molecule blind docking reveal a possible target site on the SARS-CoV-2 main protease dimer interface. Comput. Biol. Chem. 2020, 89, 107372. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [Green Version]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tufféry, P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef] [Green Version]
- Bruno, C.; Cavalluzzi, M.M.; Rusciano, M.R.; Lovece, A.; Carrieri, A.; Pracella, R.; Giannuzzi, G.; Polimeno, L.; Viale, M.; Illario, M. The chemosensitizing agent lubeluzole binds calmodulin and inhibits Ca2+/calmodulin-dependent kinase II. Eur. J. Med. Chem. 2016, 116, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, W.; Liu, S.; Xu, J. RaptorX-Property: A web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalidas, Y.; Chandra, N. PocketDepth: A new depth based algorithm for identification of ligand binding sites in proteins. J. Struct. Biol. 2008, 161, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.-F.; Fischer, P.D.; Coote, P.W.; Das, K.M.P.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Cheminform. 2012, 4, P17. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Swamidass, S.J.; Dou, Y.; Bruand, J.; Baldi, P. ChemDB: A public database of small molecules and related chemoinformatics resources. Bioinformatics 2005, 21, 4133–4139. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today: Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Whitty, A.; Zhong, M.; Viarengo, L.; Beglov, D.; Hall, D.R.; Vajda, S. Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discov. Today 2016, 21, 712–717. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Robertson, D.H.; Brooks, C.L., III; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins: Struct. Funct. Bioinform. 1999, 37, 228–241. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, M.; Fielding, B.; Gamieldien, J. Practical considerations in virtual screening and molecular docking. Emerg. Trends Comput. Biol. Bioinform. Syst. Biol. 2015, 1762, 487–502. [Google Scholar] [CrossRef]
- Yadava, U. Search algorithms and scoring methods in protein-ligand docking. Endocrinol. Metab. Int. J. 2018, 6, 359–367. [Google Scholar] [CrossRef]
- Hart, T.N.; Read, R.J. A multiple-start Monte Carlo docking method. Proteins: Struct. Funct. Bioinform. 1992, 13, 206–222. [Google Scholar] [CrossRef]
- Oshiro, C.M.; Kuntz, I.D.; Dixon, J.S. Flexible ligand docking using a genetic algorithm. J. Comput. Aided Mol. Des. 1995, 9, 113–130. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdiscip. Sci.: Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Stärk, H.; Ganea, O.; Pattanaik, L.; Barzilay, R.; Jaakkola, T. Equibind: Geometric deep learning for drug binding structure prediction. In Proceedings of the International Conference on Machine Learning, Baltimore, MD, USA, 17–23 July 2022; pp. 20503–20521. [Google Scholar]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular docking with deep learning. J. Cheminformatics 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Stärk, H.; Jing, B.; Barzilay, R.; Jaakkola, T. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking. arXiv 2022, arXiv:2210.01776. [Google Scholar]
- Salsbury, F.R., Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Philip, P.; Anshuman, D.; Anil, K.S. Computer-aided drug design: Integration of structure-based and ligand-based approaches in drug design. Curr. Comput. Aided Drug Des. 2007, 3, 133–148. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef] [Green Version]
- Van Drie, J.H. Generation of three-dimensional pharmacophore models. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2013, 3, 449–464. [Google Scholar] [CrossRef]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Barnum, D.; Greene, J.; Smellie, A.; Sprague, P. Identification of common functional configurations among molecules. J. Chem. Inf. Comput. Sci. 1996, 36, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Chen, I.-J.; Foloppe, N. Conformational sampling of druglike molecules with MOE and catalyst: Implications for pharmacophore modeling and virtual screening. J. Chem. Inf. Model. 2008, 48, 1773–1791. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Mallik, B.; Morikis, D. Development of a quasi-dynamic pharmacophore model for anti-complement peptide analogues. J. Am. Chem. Soc. 2005, 127, 10967–10976. [Google Scholar] [CrossRef]
- Langer, T.; Wolber, G. Pharmacophore definition and 3D searches. Drug Discov. Today: Technol. 2004, 1, 203–207. [Google Scholar] [CrossRef]
- Melo-Filho, C.C.; Braga, R.C.; Andrade, C.H. 3D-QSAR approaches in drug design: Perspectives to generate reliable CoMFA models. Curr. Comput. Aided Drug Des. 2014, 10, 148–159. [Google Scholar] [CrossRef]
- Sydow, D. Dynophores: Novel Dynamic Pharmacophores; Humboldt-Universität zu Berlin, Lebenswissenschaftliche Fakultät: Berlin, Germany, 2015. [Google Scholar]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design—A review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.M.; Noolvi, M.N.; Sharma, P.; Jaiswal, V.; Bansal, S.; Lohan, S.; Kumar, S.S.; Abbot, V.; Dhiman, S.; Bhardwaj, V. Quantitative structure–activity relationship (QSAR) studies as strategic approach in drug discovery. Med. Chem. Res. 2014, 23, 4991–5007. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Wang, F.; Shi, X.-X.; Jia, C.-Y.; Wu, F.-X.; Hao, G.-F.; Yang, G.-F. Cloud 3D-QSAR: A web tool for the development of quantitative structure–activity relationship models in drug discovery. Brief. Bioinform. 2020, 22, bbaa276. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.P.A.; de Oliveira, M.A.R.; de Queiroz, M.S.O. Web-4D-QSAR: A web-based application to generate 4D-QSAR descriptors. J. Comput. Chem. 2018, 39, 917–924. [Google Scholar] [CrossRef]
- Soufan, O.; Ba-Alawi, W.; Magana-Mora, A.; Essack, M.; Bajic, V.B. DPubChem: A web tool for QSAR modeling and high-throughput virtual screening. Sci. Rep. 2018, 8, 9110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, C.; Wang, F. MoFlow: An invertible flow model for generating molecular graphs. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, San Diego, CA, USA, 6–10 July 2020; pp. 617–626. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proceedings of the 35th International Conference on MachineLearning, Stockholm, Sweden, 10–15 July 2018; pp. 2323–2332. [Google Scholar]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Z.; Sun, H.; Wang, J.; Shen, C.; Weng, G.; Chai, X.; Li, H.; Cao, D.; Hou, T. Deep learning approaches for de novo drug design: An overview. Curr. Opin. Struct. Biol. 2022, 72, 135–144. [Google Scholar] [CrossRef]
- Chenthamarakshan, V.; Das, P.; Hoffman, S.; Strobelt, H.; Padhi, I.; Lim, K.W.; Hoover, B.; Manica, M.; Born, J.; Laino, T. CogMol: Target-specific and selective drug design for COVID-19 using deep generative models. Adv. Neural Inf. Process. Syst. 2020, 33, 4320–4332. [Google Scholar]
- Krishnan, S.R.; Bung, N.; Bulusu, G.; Roy, A. Accelerating de novo drug design against novel proteins using deep learning. J. Chem. Inf. Model. 2021, 61, 621–630. [Google Scholar] [CrossRef]
- Coley, C.W.; Green, W.H.; Jensen, K.F. Machine learning in computer-aided synthesis planning. Acc. Chem. Res. 2018, 51, 1281–1289. [Google Scholar] [CrossRef]
- Luo, Y.; Yan, K.; Ji, S. Graphdf: A discrete flow model for molecular graph generation. In Proceedings of the 38th International Conference on Machine Learning, Online, 18–24 July 2021; pp. 7192–7203. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Hierarchical generation of molecular graphs using structural motifs. In Proceedings of the 37th International Conference on MachineLearning, Online, 13–18 July 2020; pp. 4839–4848. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Multi-objective molecule generation using interpretable substructures. In Proceedings of the 37th International Conference on MachineLearning, Online, 13–18 July 2020; pp. 4849–4859. [Google Scholar]
- Li, Y.; Hu, J.; Wang, Y.; Zhou, J.; Zhang, L.; Liu, Z. Deepscaffold: A comprehensive tool for scaffold-based de novo drug discovery using deep learning. J. Chem. Inf. Model. 2019, 60, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, S.; Guan, J.; Ma, J.; Peng, J. A 3D generative model for structure-based drug design. Adv. Neural Inf. Process. Syst. 2021, 34, 6229–6239. [Google Scholar]
- Pandey, M.; Fernandez, M.; Gentile, F.; Isayev, O.; Tropsha, A.; Stern, A.C.; Cherkasov, A. The transformational role of GPU computing and deep learning in drug discovery. Nat. Mach. Intell. 2022, 4, 211–221. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Liu, X.; Tan, X.; Li, X.; Jiang, J.; Xiong, Z.; Xu, T.; Jiang, H.; Qiao, N.; Zheng, M. Generative models for De Novo drug design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef]
- Kuznetsov, M.; Polykovskiy, D. MolGrow: A graph normalizing flow for hierarchical molecular generation. In Proceedings of the AAAI Conference on Artificial Intelligence, Online, 2–9 February 2021; pp. 8226–8234. [Google Scholar]
- Bilodeau, C.; Jin, W.; Jaakkola, T.; Barzilay, R.; Jensen, K.F. Generative models for molecular discovery: Recent advances and challenges. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2022, 12, e1608. [Google Scholar] [CrossRef]
- Adams, K.; Coley, C.W. Equivariant Shape-Conditioned Generation of 3D Molecules for Ligand-Based Drug Design. arXiv 2022, arXiv:2210.04893. [Google Scholar]
- Batool, M.; Ahmad, B.; Choi, S. A structure-based drug discovery paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef]
- Sattarov, B.; Baskin, I.I.; Horvath, D.; Marcou, G.G.; Bjerrum, E.J.; Varnek, A. De novo molecular design by combining deep autoencoder recurrent neural networks with generative topographic mapping. J. Chem. Inf. Model. 2019, 59, 1182–1196. [Google Scholar] [CrossRef]
- Li, Y.; Pei, J.; Lai, L. Structure-based de novo drug design using 3D deep generative models. Chem. Sci. 2021, 12, 13664–13675. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hsieh, C.-Y.; Wang, J.; Wang, D.; Weng, G.; Shen, C.; Yao, X.; Bing, Z.; Li, H.; Cao, D. RELATION: A Deep Generative Model for Structure-Based De Novo Drug Design. J. Med. Chem. 2022, 65, 9478–9492. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Sun, H.; Wang, J.; Pang, J.; Chai, X.; Xu, L.; Li, H.; Cao, D.; Hou, T. Comprehensive assessment of deep generative architectures for de novo drug design. Brief. Bioinform. 2022, 23, bbab544. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Bung, N.; Vangala, S.R.; Srinivasan, R.; Bulusu, G.; Roy, A. De novo structure-based drug design using deep learning. J. Chem. Inf. Model. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, N.W.A.; Gastegger, M.; Hessmann, S.S.P.; Müller, K.-R.; Schütt, K.T. Inverse design of 3d molecular structures with conditional generative neural networks. Nat. Commun. 2022, 13, 973. [Google Scholar] [CrossRef]
- Peng, X.; Luo, S.; Guan, J.; Xie, Q.; Peng, J.; Ma, J. Pocket2Mol: Efficient Molecular Sampling Based on 3D Protein Pockets. arXiv 2022, arXiv:2205.07249. [Google Scholar]
- Xie, W.; Wang, F.; Li, Y.; Lai, L.; Pei, J. Advances and Challenges in De Novo Drug Design Using Three-Dimensional Deep Generative Models. J. Chem. Inf. Model. 2022, 62, 2269–2279. [Google Scholar] [CrossRef]
- Thomas, M.; Smith, R.T.; O’Boyle, N.M.; De Graaf, C.; Bender, A. Comparison of structure-and ligand-based scoring functions for deep generative models: A GPCR case study. J. Cheminformatics 2021, 13, 39. [Google Scholar] [CrossRef]
- Bengio, E.; Jain, M.; Korablyov, M.; Precup, D.; Bengio, Y. Flow network based generative models for non-iterative diverse candidate generation. Adv. Neural Inf. Process. Syst. 2021, 34, 27381–27394. [Google Scholar] [CrossRef]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- Chen, H. Can Generative-Model-Based Drug Design Become a New Normal in Drug Discovery? J. Med. Chem. 2021, 65, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Lewell, X.Q.; Judd, D.B.; Watson, S.P.; Hann, M.M. Recap retrosynthetic combinatorial analysis procedure: A powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J. Chem. Inf. Comput. Sci. 1998, 38, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Vinkers, H.M.; de Jonge, M.; Daeyaert, F.F.; Heeres, J.; Koymans, L.M.; van Lenthe, J.H.; Lewi, P.J.; Timmerman, H.; Van Aken, K.; Janssen, P.A. SYNOPSIS: SYNthesize and OPtimize system in silico. J. Med. Chem. 2003, 46, 2765–2773. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; Von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, Q.; Yu, G.; Gai, W.; Li, Y.; Chen, X. DCDB 2.0: A major update of the drug combination database. Database 2014, 2014, bau124. [Google Scholar] [CrossRef] [PubMed]
- Fink, T.; Reymond, J.-L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: Assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model. 2007, 47, 342–353. [Google Scholar] [CrossRef]
- Blum, L.C.; Reymond, J.-L. 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 2009, 131, 8732–8733. [Google Scholar] [CrossRef] [PubMed]
- Ruddigkeit, L.; Van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, K.; Kim, W.; Jung, S.; Cho, A.E. Target-specific drug design method combining deep learning and water pharmacophore. J. Chem. Inf. Model. 2020, 61, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de novo drug design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Gupta, A.; Müller, A.T.; Huisman, B.J.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative recurrent networks for de novo drug design. Mol. Inf. 2018, 37, 1700111. [Google Scholar] [CrossRef] [Green Version]
- Arús-Pous, J.; Patronov, A.; Bjerrum, E.J.; Tyrchan, C.; Reymond, J.-L.; Chen, H.; Engkvist, O. SMILES-based deep generative scaffold decorator for de-novo drug design. J. Cheminform. 2020, 12, 38. [Google Scholar] [CrossRef]
- Liu, X.; IJzerman, A.P.; van Westen, G.J. Computational approaches for de novo drug design: Past, present, and future. Artif. Neural Netw. 2021, 139–165. [Google Scholar]
- Mandhana, V.; Taware, R. De novo drug design using self attention mechanism. In Proceedings of the 35th Annual ACM Symposium on Applied Computing, Online, 30 March–3 April 2020; pp. 8–12. [Google Scholar]
- Graves, A. Long short-term memory. In Supervised Sequence Labelling with Recurrent Neural Networks; Springer: Berlin/Heidelberg, Germany, 2012; pp. 37–45. [Google Scholar] [CrossRef]
- Yasonik, J. Multiobjective de novo drug design with recurrent neural networks and nondominated sorting. J. Cheminform. 2020, 12, 14. [Google Scholar] [CrossRef]
- Arús-Pous, J.; Johansson, S.V.; Prykhodko, O.; Bjerrum, E.J.; Tyrchan, C.; Reymond, J.-L.; Chen, H.; Engkvist, O. Randomized SMILES strings improve the quality of molecular generative models. J. Cheminform. 2019, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Van Merriënboer, B.; Bahdanau, D.; Bengio, Y. On the properties of neural machine translation: Encoder-decoder approaches. arXiv 2014, arXiv:1409.1259. [Google Scholar]
- Zheng, S.; Yan, X.; Gu, Q.; Yang, Y.; Du, Y.; Lu, Y.; Xu, J. QBMG: Quasi-biogenic molecule generator with deep recurrent neural network. J. Cheminform. 2019, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barshatski, G.; Radinsky, K. Unpaired Generative Molecule-to-Molecule Translation for Lead Optimization. In Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining, Online, 14–18 August 2021; pp. 2554–2564. [Google Scholar]
- Rezaei, M.A.; Li, Y.; Wu, D.; Li, X.; Li, C. Deep learning in drug design: Protein-ligand binding affinity prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Francoeur, P.G.; Masuda, T.; Sunseri, J.; Jia, A.; Iovanisci, R.B.; Snyder, I.; Koes, D.R. Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inf. Model. 2020, 60, 4200–4215. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; pp. 1–11. [Google Scholar]
- Cheng, Z.; Yan, C.; Wu, F.-X.; Wang, J. Drug-target interaction prediction using multi-head self-attention and graph attention network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 19, 2208–2218. [Google Scholar] [CrossRef]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural message passing for quantum chemistry. In Proceedings of Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 1263–1272. [Google Scholar]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 2019, 63, 8749–8760. [Google Scholar] [CrossRef]
- Griffiths, R.-R.; Hernández-Lobato, J.M. Constrained Bayesian optimization for automatic chemical design using variational autoencoders. Chem. Sci. 2020, 11, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Asperti, A.; Trentin, M. Balancing reconstruction error and Kullback-Leibler divergence in Variational Autoencoders. IEEE Access 2020, 8, 199440–199448. [Google Scholar] [CrossRef]
- Wei, R.; Mahmood, A. Recent advances in variational autoencoders with representation learning for biomedical informatics: A survey. IEEE Access 2020, 9, 4939–4956. [Google Scholar] [CrossRef]
- Liu, Q.; Allamanis, M.; Brockschmidt, M.; Gaunt, A. Constrained graph variational autoencoders for molecule design. In Proceedings of the 32nd Conference on Neural Information Processing Systems (NeurIPS 2018), Montréal, QC, Canada, 3–8 December 2018. [Google Scholar]
- Simonovsky, M.; Komodakis, N. Graphvae: Towards generation of small graphs using variational autoencoders. In Proceedings of the International Conference on Artificial Neural Networks, Rhodes, Greece, 4–7 October 2018; pp. 412–422. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. Commun. ACM 2020, 63, 139–144. [Google Scholar] [CrossRef]
- De Cao, N.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar]
- Madhawa, K.; Ishiguro, K.; Nakago, K.; Abe, M. Graphnvp: An invertible flow model for generating molecular graphs. arXiv 2019, arXiv:1905.11600. [Google Scholar]
- Papamakarios, G.; Pavlakou, T.; Murray, I. Masked autoregressive flow for density estimation. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; pp. 1–10. [Google Scholar]
- Xiong, J.; Xiong, Z.; Chen, K.; Jiang, H.; Zheng, M. Graph neural networks for automated de novo drug design. Drug Discov. Today 2021, 26, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Stärk, H.; Beaini, D.; Corso, G.; Tossou, P.; Dallago, C.; Günnemann, S.; Liò, P. 3D infomax improves gnns for molecular property prediction. In Proceedings of the International Conference on Machine Learning, Baltimore, MD, USA, 17–23 July 2022; pp. 20479–20502. [Google Scholar]
- Fabian, B.; Edlich, T.; Gaspar, H.; Segler, M.; Meyers, J.; Fiscato, M.; Ahmed, M. Molecular representation learning with language models and domain-relevant auxiliary tasks. arXiv 2020, arXiv:2011.13230. [Google Scholar]
- Wang, S.; Guo, Y.; Wang, Y.; Sun, H.; Huang, J. SMILES-BERT: Large scale unsupervised pre-training for molecular property prediction. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; pp. 429–436. [Google Scholar]
- Reidenbach, D.; Livne, M.; Ilango, R.K.; Gill, M.; Israeli, J. Improving Small Molecule Generation using Mutual Information Machine. arXiv 2022, arXiv:2208.09016. [Google Scholar]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-scale self-supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Edwards, C.; Lai, T.; Ros, K.; Honke, G.; Ji, H. Translation between Molecules and Natural Language. arXiv 2022, arXiv:2204.11817. [Google Scholar]
- Chen, Z.; Min, M.R.; Parthasarathy, S.; Ning, X. A deep generative model for molecule optimization via one fragment modification. Nat. Mach. Intell. 2021, 3, 1040–1049. [Google Scholar] [CrossRef]
- Button, A.; Merk, D.; Hiss, J.A.; Schneider, G. Automated de novo molecular design by hybrid machine intelligence and rule-driven chemical synthesis. Nat. Mach. Intell. 2019, 1, 307–315. [Google Scholar] [CrossRef]
- Chen, B.; Wang, T.; Li, C.; Dai, H.; Song, L. Molecule optimization by explainable evolution. In Proceedings of the International Conference on Learning Representation (ICLR 2021), Online, 3–7 May 2021; pp. 1–15. [Google Scholar]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep docking: A deep learning platform for augmentation of structure based drug discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Engkvist, O. Has drug design augmented by artificial intelligence become a reality? Trends Pharmacol. Sci. 2019, 40, 806–809. [Google Scholar] [CrossRef]
- Klebe, G. On the validity of popular assumptions in computational drug design. J. Cheminform. 2011, 3, O18. [Google Scholar] [CrossRef] [Green Version]
- Garrido, A.; Lepailleur, A.; Mignani, S.M.; Dallemagne, P.; Rochais, C. hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem. 2020, 195, 112290. [Google Scholar] [CrossRef] [PubMed]
- Hessler, G.; Baringhaus, K.-H. Artificial intelligence in drug design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Luo, Y.; Uchino, K.; Maruhashi, K.; Ji, S. Generating 3D Molecules for Target Protein Binding. arXiv 2022, arXiv:2204.09410. [Google Scholar]
- Xie, Y.; Shi, C.; Zhou, H.; Yang, Y.; Zhang, W.; Yu, Y.; Li, L. Mars: Markov molecular sampling for multi-objective drug discovery. arXiv 2021, arXiv:2103.10432. [Google Scholar]
- Eckmann, P.; Sun, K.; Zhao, B.; Feng, M.; Gilson, M.K.; Yu, R. LIMO: Latent Inceptionism for Targeted Molecule Generation. arXiv 2022, arXiv:2206.09010. [Google Scholar]
- Renz, P.; Van Rompaey, D.; Wegner, J.K.; Hochreiter, S.; Klambauer, G. On failure modes in molecule generation and optimization. Drug Discov. Today: Technol. 2019, 32–33, 55–63. [Google Scholar] [CrossRef]
- Battaglia, P.W.; Hamrick, J.B.; Bapst, V.; Sanchez-Gonzalez, A.; Zambaldi, V.; Malinowski, M.; Tacchetti, A.; Raposo, D.; Santoro, A.; Faulkner, R. Relational inductive biases, deep learning, and graph networks. arXiv 2018, arXiv:1806.01261. [Google Scholar]
- Atz, K.; Grisoni, F.; Schneider, G. Geometric deep learning on molecular representations. Nat. Mach. Intell. 2021, 3, 1023–1032. [Google Scholar] [CrossRef]
- Flam-Shepherd, D.; Zhu, K.; Aspuru-Guzik, A. Language models can learn complex molecular distributions. Nat. Commun. 2022, 13, 3293. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wu, Q.; Zhang, J.; Rao, J.; Li, C.; Zheng, S. TANKBind: Trigonometry-Aware Neural NetworKs for Drug-Protein Binding Structure Prediction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zhou, G.; Gao, Z.; Ding, Q.; Zheng, H.; Xu, H.; Wei, Z.; Zhang, L.; Ke, G. Uni-Mol: A Universal 3D Molecular Representation Learning Framework. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, B. Computing absolute free energy with deep generative models. J. Phys. Chem. B 2020, 124, 10166–10172. [Google Scholar] [CrossRef]
- Lahey, S.L.J.; Rowley, C.N. Simulating protein–ligand binding with neural network potentials. Chem. Sci. 2020, 11, 2362–2368. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Han, J.; Wang, H.; Car, R.; Weinan, E. Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 2018, 120, 143001. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Feng, H.; Wu, J.; Xia, K. Persistent spectral hypergraph based machine learning (PSH-ML) for protein-ligand binding affinity prediction. Brief. Bioinform. 2021, 22, bbab127. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, T.B.; Dahlgren, M.K.; Cappel, D.; Lin, T.; Wang, L.; Krilov, G.; Abel, R.A.; Friesner, R.; Sherman, W. Accurate binding free energy predictions in fragment optimization. J. Chem. Inf. Model. 2015, 55, 2411–2420. [Google Scholar] [CrossRef]
- Tayara, H.; Abdelbaky, I.; Chong, K.T. Recent omics-based computational methods for COVID-19 drug discovery and repurposing. Brief. Bioinform. 2021, 22, bbab339. [Google Scholar] [CrossRef]








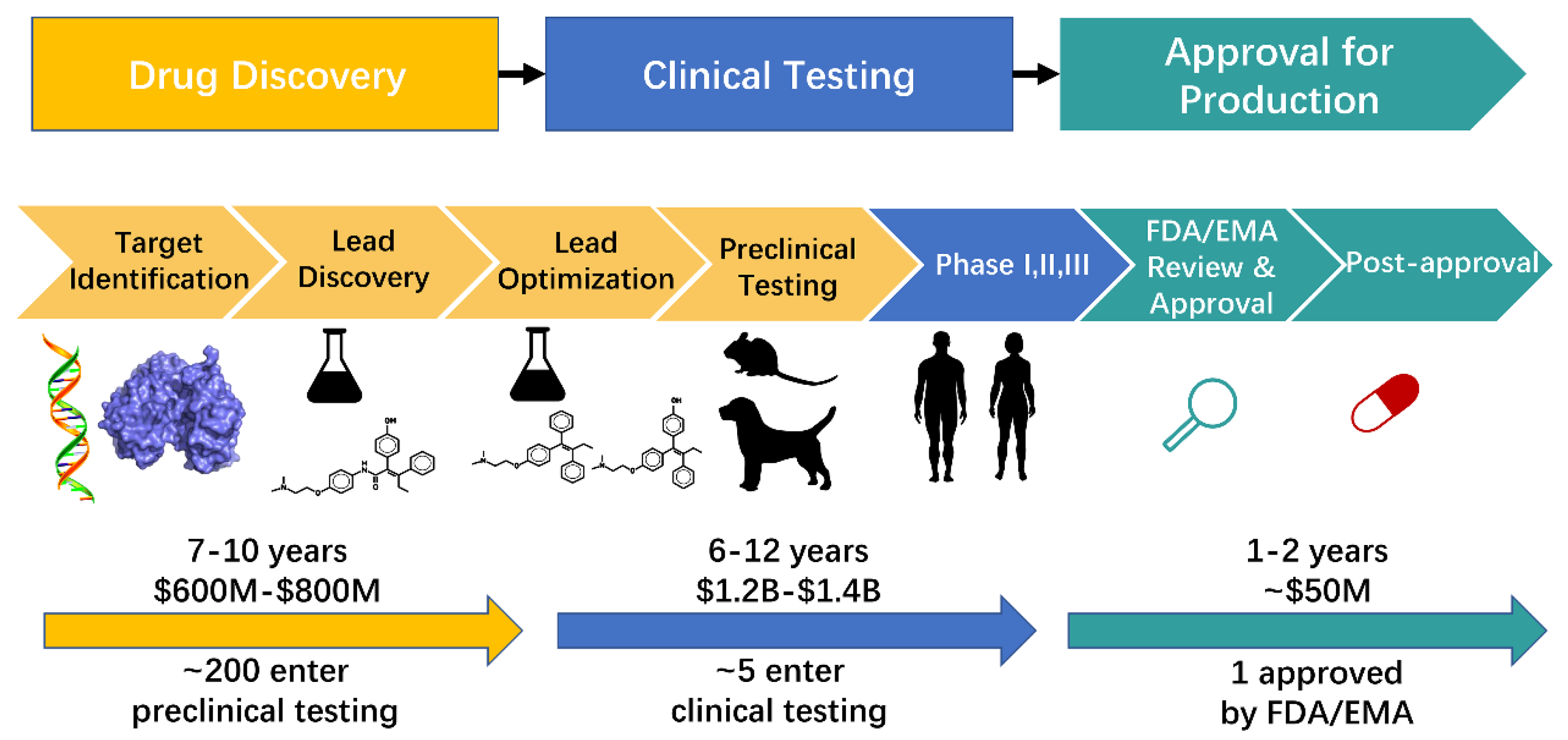{kind=link}
{kind=link}
{kind=link}
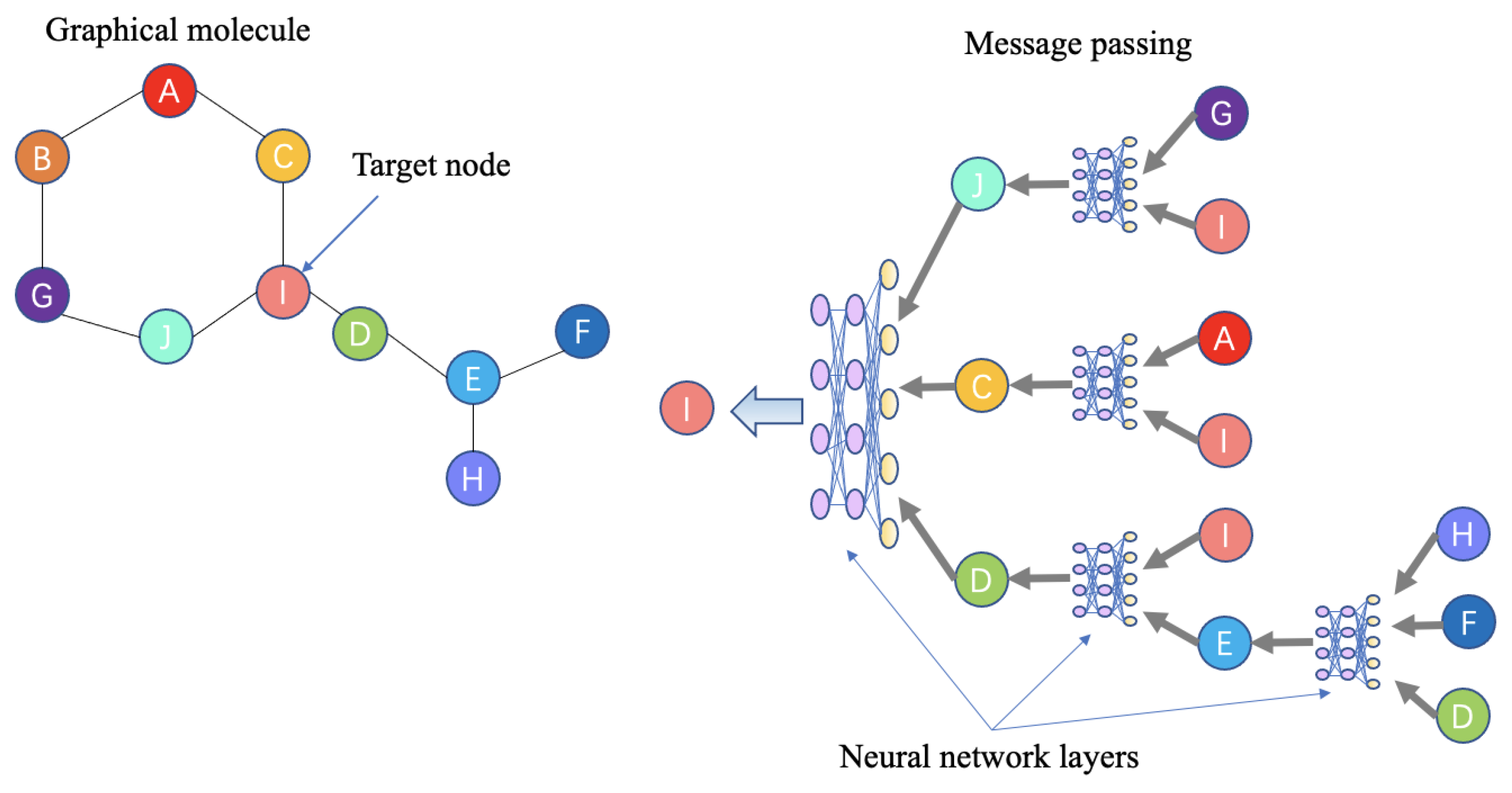{kind=link}
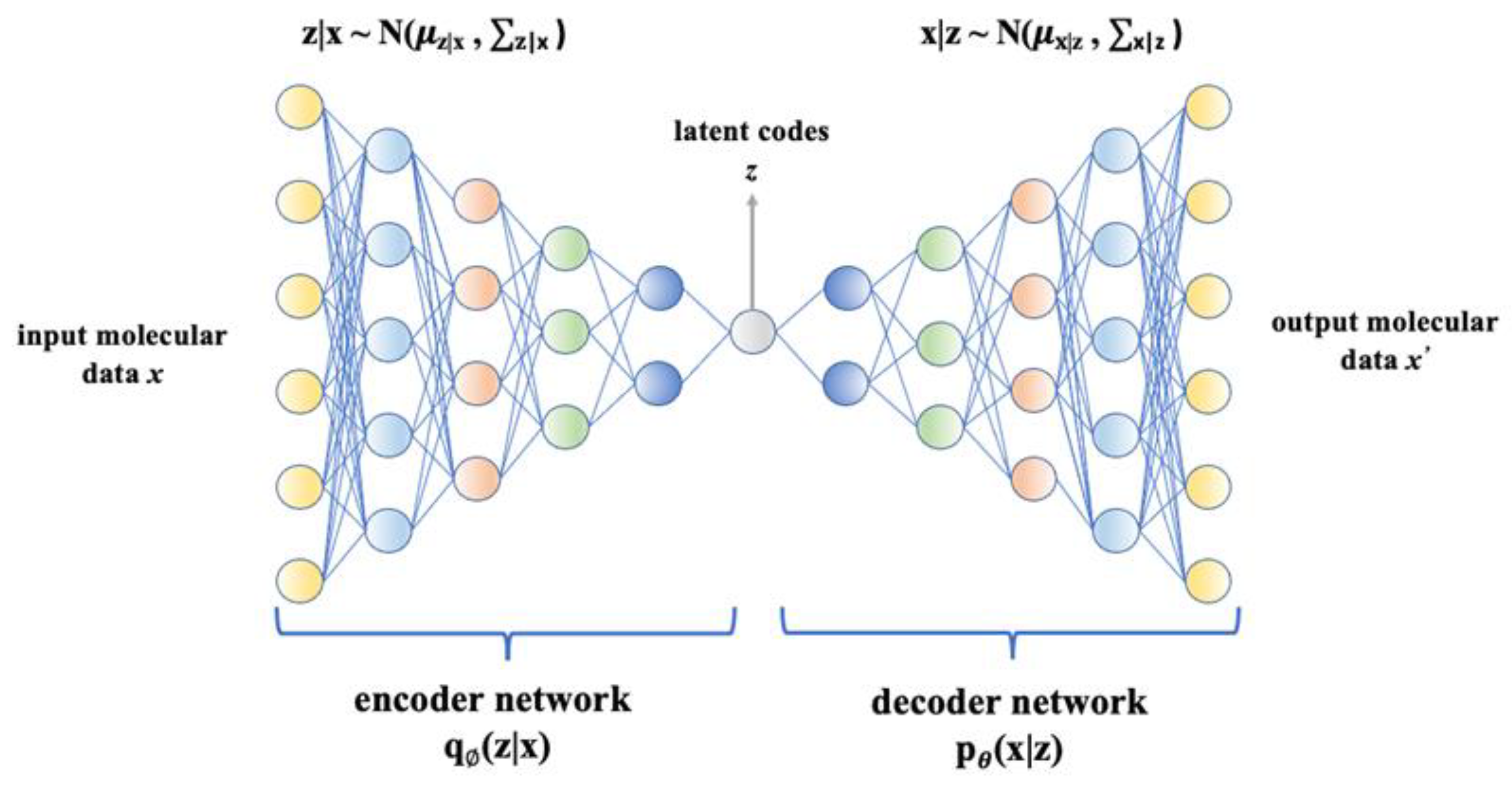{kind=link}
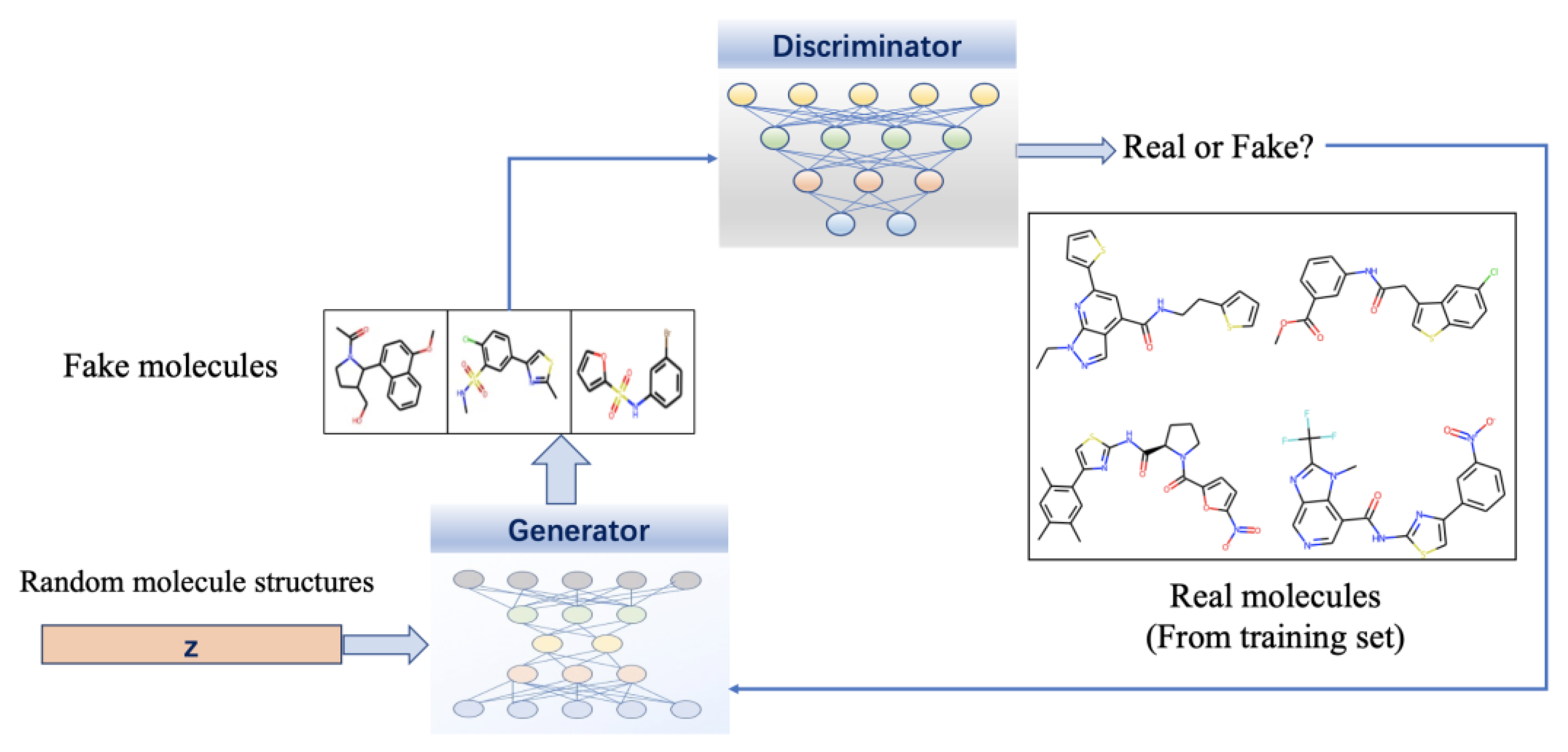{kind=link}
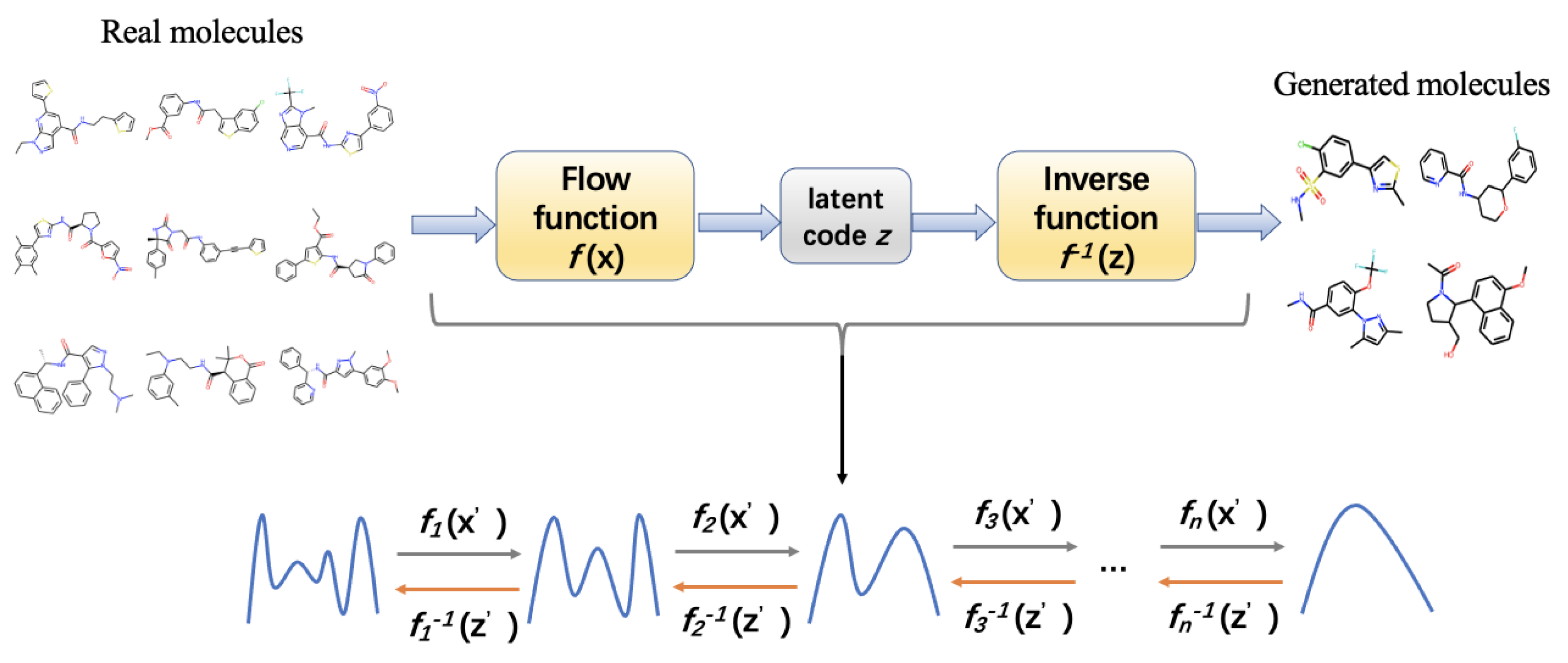{kind=link}
| Compound Database | Description | Number of Compounds |
|---|---|---|
| ChEMBL [264,265] | Drug discovery database provides bioactive molecules with drug-like properties knowledge. | 2,157,379 |
| ZINC [178,231,232,233,254] | Database enables access to compounds for drug discovery. | 750 million + 230 million (3D) |
| PubChem [233,264,266] | Public chemical database at the National Library of Medicine (NLM) collects chemical information from different data sources. | 112 million |
| DrugBank [183,231,232,254,264] | Web resource contains drug-related information. | 14,528 |
| STITCH [264,267] | Database contains interaction information between different chemicals. | 0.5 million |
| BindingDB [264,268] | Database for molecular recognition, which supports drug discovery related work. | 1.1 million |
| SIDER [264,269] | Resource contains drug reactions information. | 1430 + 55,730 |
| DCDB [264,270] | Drug Combination Database | 1363 |
| GDB-11 [231,232,254,271] | Database collects and generates molecules with up to 11 atoms of C, N, O, and F by considering simple valency, chemical stability, and synthetic feasibility rules. | 26.4 million |
| GDB-13 [231,232,254,272] | Database upgrading from GDB-11, it enumerates in a similar manner small organic molecules containing up to 13 atoms of C, N, O, S, and Cl. | 970 million |
| GDB-17 [231,232,254,273] | Chemical universe database covers drugs and typical for lead compounds for molecules with up to 17 atoms of C, N, O, S, and halogens. | 166 billion |
| Deep Learning Techniques | Description | Applications |
|---|---|---|
| Recurrent neural networks (RNN) | Recurrent neural networks are similar to Markov chains with memory and feedback loops, each neuron in it would receive information from both actual time input and the previous neural [232,241,264] | SMILES strings representation [234,241,243]; generating novel and valid SMILES strings [34]; learn model autoregression [241]; construct encoder to convert discrete representations of molecules to multidimensional continuous representation [231]; estimate the probabilities of molecular data [259] |
| Long Short-Term Memory (LSTM) | LSTM is one kind of recurrent neural work with attention mechanism, which aims to solve the vanishing gradient problem for Recurrent Neural Networks (RNNs) [280,281] | encode SMILES strings [282]; builds sequence-to-sequence neural network for autoencoder [249] |
| Gated Recurrent Neural Network (Gated RNN) | One type of RNN with gated recurrent unit (GRU) containing forget gate and updating gate [283,284] | constructing encoder and decoder for SMILES sequence translation [253,285]; junction tree message passing [230] |
| Convolutional neural networks (CNN) | Convolutional neural networks contain sequential layers of convolution and pooling, and among them the convolution layers extract features by moving a window over the input tensors (arrays) and the pooling layers sub-sample the features [241,264,286] | construct graph convolutional neural networks [239]; grid-based 3D CNNs to predict protein–ligand binding affinity by constructing [287] |
| Multilayer perceptron networks (MPL) | Deep neural networks consist of multilayer perceptions, which are fully connected networks with activation functions [241] | chemical properties from latent codes [231]; mapping between latent vectors and molecular properties [229] |
| Multi-head attention networks | Contains encoder and decoder both with stacked self-attention and fully-connected layer inside, and the attention blocks in the network are all in the form of multi-head for receiving inputs of query, key, and value [288,289] | extract 3D conditional information of molecule [236] embed the active site graphs of target [253] |
| Message passing neural networks (MPNN) | A state-of-the-art and typical model for learning nodes and edges information in graph: a target node’s representation come from its directly connected nodes through a multilayer neural network (or one layer), and the message passing between nodes in the graph is a circular iteration process [290]. | parameterize atom graph encoding [243] encode connected motifs information of molecule [237] graph message passing network to represent the junction tree and molecular graph into latent codes [193] learn molecular graph and rationale distribution [238] |
| Graph neural network (GNN) | Regarding atoms as nodes and bonds as edges, this network applies convoluting operations for graphs encoding [40,264] | atoms and bonds information representation [240,245] parameterized the encoder and decoder for atoms and bonds types [239] spherical message passing graph neural networks to extract 3D conditional information of molecule [236] |
| Evaluation Metrics | Descriptions |
|---|---|
| LogP | The oil-water partition coefficient, also called the hydrophobic constant; the larger the LogP value, the more lipophilic the drug is; conversely, the smaller the LogP value, the more hydrophilic the drug is [233,239,243,245]. |
| QED | Quantitative estimate of drug-likeness, and the value it is between 0 and 1 [239,240,250,309]. |
| Synthesizability | The probability of the generated drug to be synthesized [233,240,277,309]. |
| Binding affinity | The magnitude of the interaction force between receptor and ligand. It can be expressed by free binding energy [240,253]. |
| Diversity | Generated molecules are similar in terms of the desired properties but with variety of forms [237,238,239,240,245]. |
| Maximum Mean Discrepancy | Maximum Mean Discrepancy values between generated molecules and real molecules [236,239,245,250]. |
| Docking score | To measure the probability of the mutual recognition between ligand and receptor through the matching principle [242,245,250]. |
| Novelty | The quality for generated molecules to be different from existed molecules, new and unusual [238,312]. |
| Validity | An inherent property of a drug, it represents the performance of drug in prevention, treatment, diagnosis of diseases and regulation of physiological functions [230,236,237,313]. |
| Similarity | Similarity between generated molecules and real molecules, such as Tanimoto Similarity between molecular fingerprints [233,234]. |
| Toxicity | The degree of poisonous or harmful that the drug would be [233,314,315]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Luo, M.; Wu, P.; Wu, S.; Lee, T.-Y.; Bai, C. Application of Computational Biology and Artificial Intelligence in Drug Design. Int. J. Mol. Sci. 2022, 23, 13568. https://doi.org/10.3390/ijms232113568
Zhang Y, Luo M, Wu P, Wu S, Lee T-Y, Bai C. Application of Computational Biology and Artificial Intelligence in Drug Design. International Journal of Molecular Sciences. 2022; 23(21):13568. https://doi.org/10.3390/ijms232113568
Chicago/Turabian StyleZhang, Yue, Mengqi Luo, Peng Wu, Song Wu, Tzong-Yi Lee, and Chen Bai. 2022. "Application of Computational Biology and Artificial Intelligence in Drug Design" International Journal of Molecular Sciences 23, no. 21: 13568. https://doi.org/10.3390/ijms232113568