
Comparative Transcriptome Analysis Identifies Key Defense Genes and Mechanisms in Mulberry (Morus alba) Leaves against Silkworms (Bombyx mori)

Abstract
:1. Introduction
2. Results
2.1. Transcriptome Profiling of Mulberry Plants
2.2. Functional Annotations of Unigenes
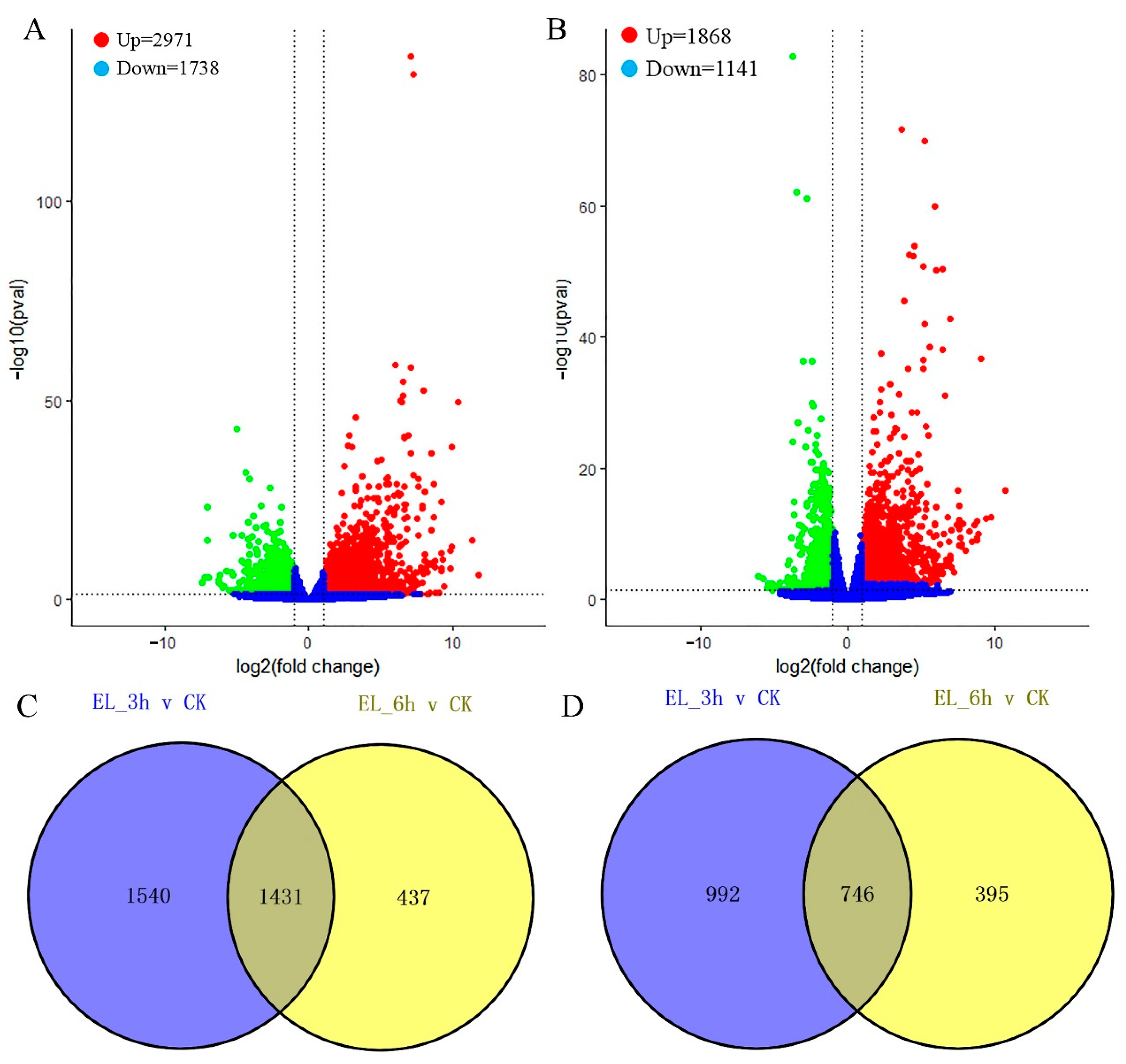
2.3. Differentially Expressed Genes (DEGs) Calculation
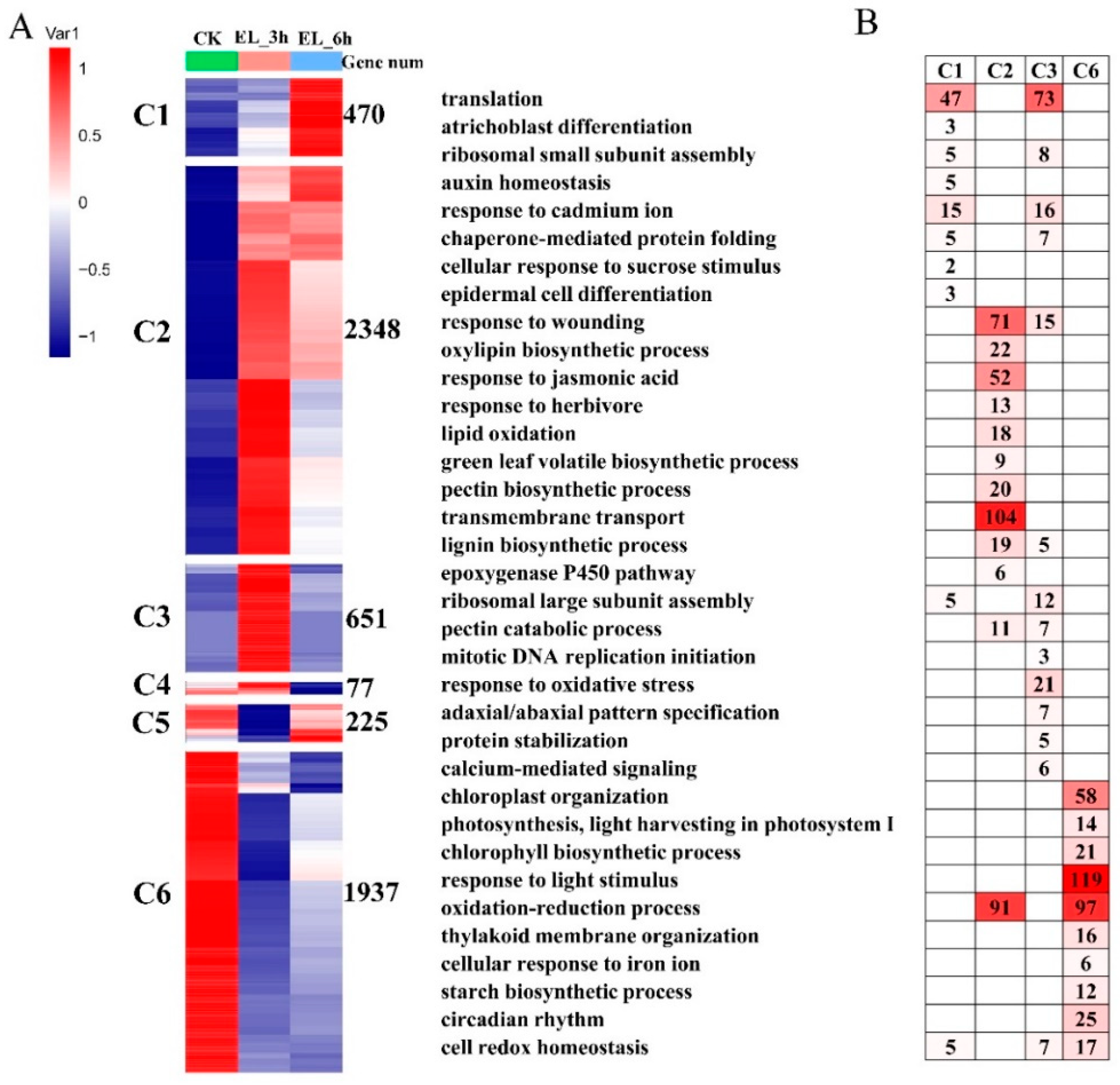
2.4. DEGs at Two Time Points and Function Enrichments
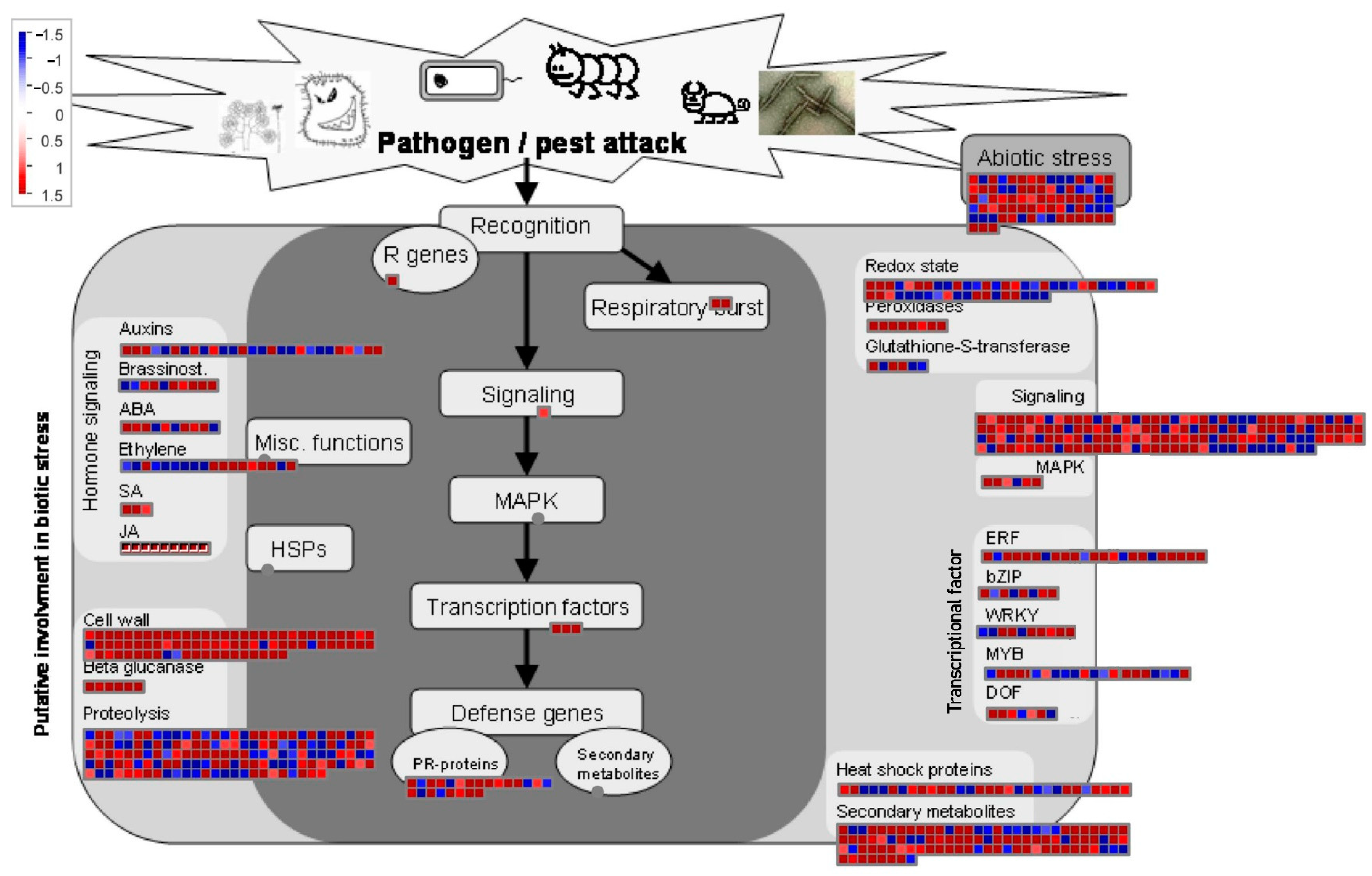
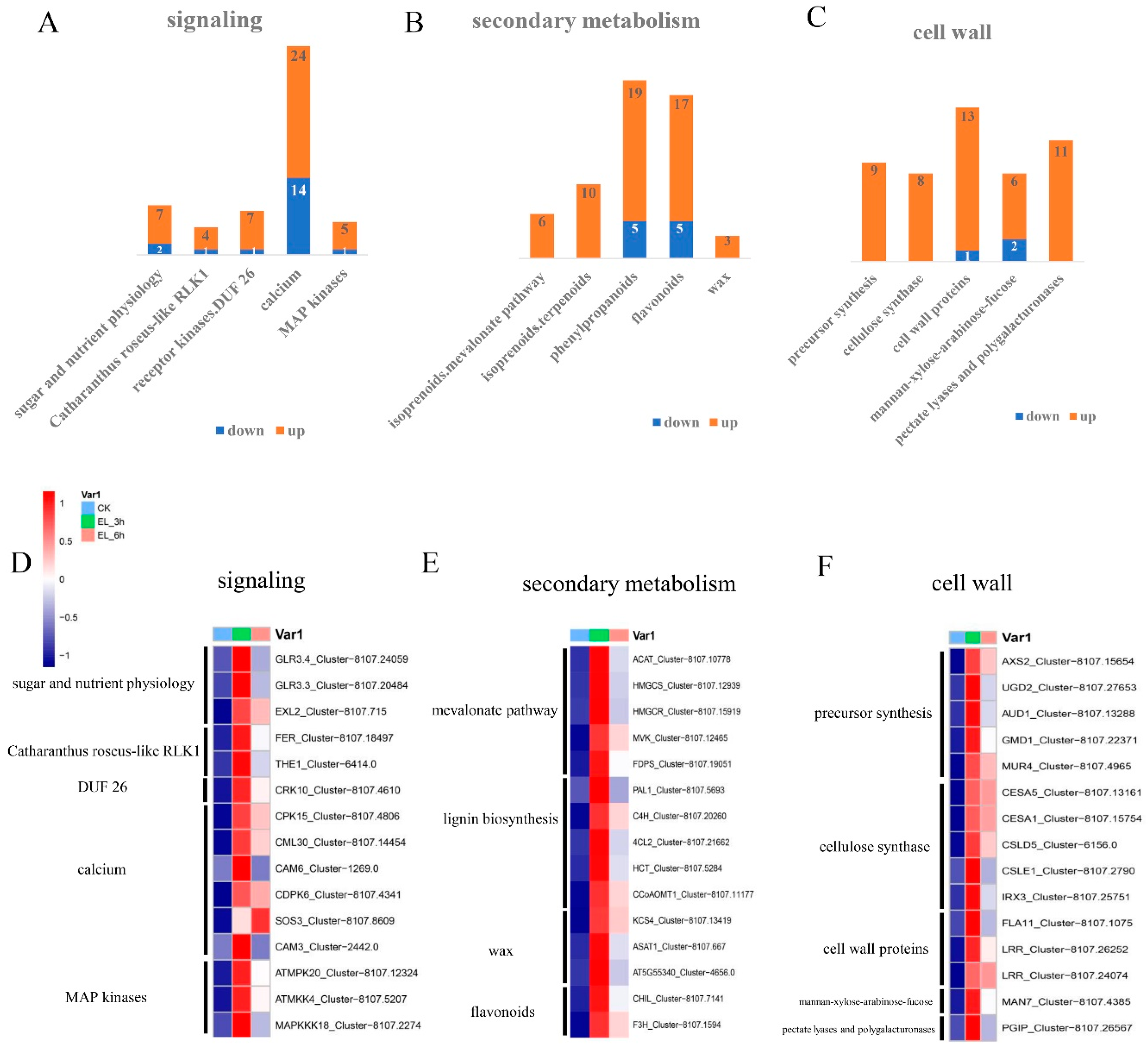
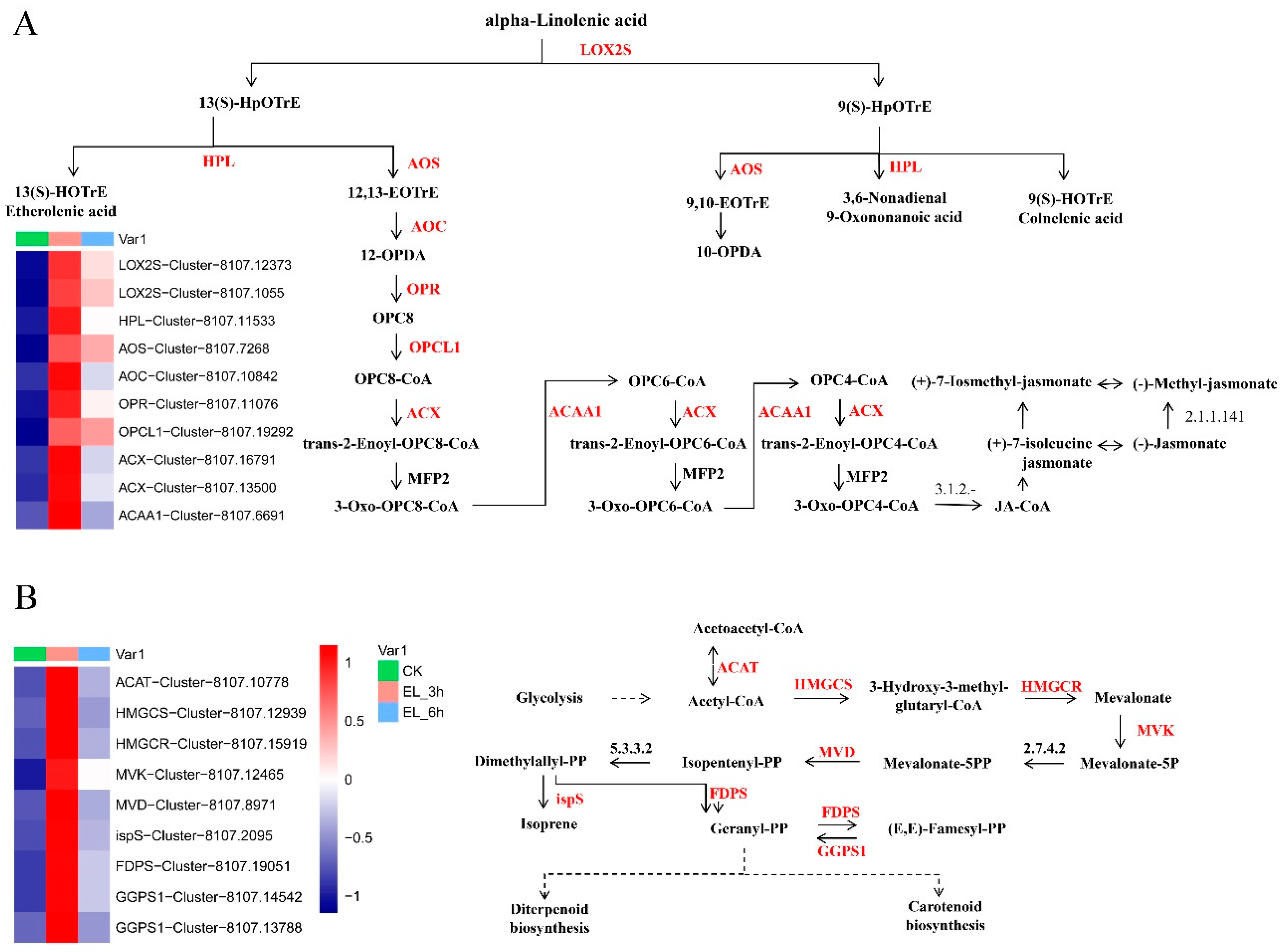
2.5. Expression Patterns of Genes of Biotic Attack Responding Pathways
2.6. Real-Time Quantitative PCR Validation
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Insect Materials and Insect Feeding Experiment
4.3. RNA Extraction
4.4. Illumina Library Construction and Sequencing
4.5. De Novo Assembly of Transcriptome
4.6. Calculation of Gene Expression
4.7. Bioinformatics Analysis
4.8. qRT-PCR Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| JA | Jasmonic acid |
| DAMPs | Damage-associated molecular patterns |
| PRRs | Pattern recognition receptors |
| ROS | Reactive oxygen species |
| MAPK | Mitogen-activated protein kinase |
| TFs | Transcription factors |
| DEGs | Differentially expressed genes |
| GO | Gene ontology |
| qRT-PCR | Real-time quantitative PCR |
| LRR-RK | Leucine-rich repeat receptor kinase |
| CaMs | Calmodulins |
| CMLs | Calmodulin-like proteins |
| CDPKs | Calcium-dependent protein kinasesBP biological processes |
References
- Li, R.; Liu, L.; Dominic, K.; Wang, T.; Fan, T.; Hu, F.; Wang, Y.; Zhang, L.; Li, L.; Zhao, W. Mulberry (Morus alba) MmSK gene enhances tolerance to drought stress in transgenic mulberry. Plant Physiol. Biochem. 2018, 132, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.; Mogili, T.; Sivaprasad, V. Improvement of abiotic stress adaptive traits in mulberry (Morus spp.): An update on biotechnological interventions. 3 Biotech 2017, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Luo, G.; Li, Z.; Wei, X.; Wang, Z.; Lin, S.; Tang, C. Physiological and transcriptomic analyses of mulberry (Morus atropurpurea) response to cadmium stress. Ecotoxicol. Environ. Saf. 2020, 205, 111298. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Chauhan, H.; Chhibbar, A.; Rizwanul Haq, Q.M.; Khurana, P. High-efficiency transformation and selective tolerance against biotic and abiotic stress in mulberry, Morus indica cv. K2, by constitutive and inducible expression of tobacco osmotin. Transgenic Res. 2011, 20, 231–246. [Google Scholar] [CrossRef]
- Dong, H.L.; Zhang, S.X.; Tao, H.; Chen, Z.H.; Li, X.; Qiu, J.F.; Cui, W.Z.; Sima, Y.H.; Cui, W.Z.; Xu, S.Q. Metabolomics differences between silkworms (Bombyx mori) reared on fresh mulberry (Morus) leaves or artificial diets. Sci. Rep. 2017, 7, 10972. [Google Scholar] [CrossRef] [Green Version]
- Qin, D.; Wang, G.; Dong, Z.; Xia, Q.; Zhao, P. Comparative Fecal Metabolomes of Silkworms Being Fed Mulberry Leaf and Artificial Diet. Insects 2020, 11, 851. [Google Scholar] [CrossRef]
- Srivastava, S.; Kapoor, R.; Thathola, A.; Srivastava, R.P. Nutritional quality of leaves of some genotypes of mulberry (Morus alba). Int. J. Food Sci. Nutr. 2006, 57, 305–313. [Google Scholar] [CrossRef]
- Erb, M.; Reymond, P. Molecular Interactions Between Plants and Insect Herbivores. Annu. Rev. Plant Biol. 2019, 70, 527–557. [Google Scholar] [CrossRef] [Green Version]
- Aljbory, Z.; Chen, M.S. Indirect plant defense against insect herbivores: A review. Insect Sci. 2018, 25, 2–23. [Google Scholar] [CrossRef]
- Acevedo, F.E.; Rivera-Vega, L.J.; Chung, S.H.; Ray, S.; Felton, G.W. Cues from chewing insects—The intersection of DAMPs, HAMPs, MAMPs and effectors. Curr. Opin. Plant Biol. 2015, 26, 80–86. [Google Scholar] [CrossRef]
- Stahl, E.; Hilfiker, O.; Reymond, P. Plant-arthropod interactions: Who is the winner? Plant J. 2018, 93, 703–728. [Google Scholar] [CrossRef] [PubMed]
- Brutus, A.; Sicilia, F.; Macone, A.; Cervone, F.; De Lorenzo, G. A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides. Proc. Natl. Acad. Sci. USA 2010, 107, 9452–9457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohorn, B.D.; Johansen, S.; Shishido, A.; Todorova, T.; Martinez, R.; Defeo, E.; Obregon, P. Pectin activation of MAP kinase and gene expression is WAK2 dependent. Plant J. 2009, 60, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaghan, J.; Zipfel, C. Plant pattern recognition receptor complexes at the plasma membrane. Curr. Opin. Plant Biol. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Scholz, S.S.; Vadassery, J.; Heyer, M.; Reichelt, M.; Bender, K.W.; Snedden, W.A.; Boland, W.; Mithofer, A. Mutation of the Arabidopsis calmodulin-like protein CML37 deregulates the jasmonate pathway and enhances susceptibility to herbivory. Mol. Plant 2014, 7, 1712–1726. [Google Scholar] [CrossRef] [Green Version]
- Monte, I.; Ishida, S.; Zamarreno, A.M.; Hamberg, M.; Franco-Zorrilla, J.M.; Garcia-Casado, G.; Gouhier-Darimont, C.; Reymond, P.; Takahashi, K.; Garcia-Mina, J.M.; et al. Ligand-receptor co-evolution shaped the jasmonate pathway in land plants. Nat. Chem. Biol. 2018, 14, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.; Schlauch, K.; Tam, R.; Cortes, D.; Torres, M.A.; Shulaev, V.; Dangl, J.L.; Mittler, R. The plant NADPH oxidase RBOHD mediates rapid systemic signaling in response to diverse stimuli. Sci. Signal. 2009, 2, ra45. [Google Scholar] [CrossRef] [Green Version]
- Block, A.; Christensen, S.A.; Hunter, C.T.; Alborn, H.T. Herbivore-derived fatty-acid amides elicit reactive oxygen species burst in plants. J. Exp. Bot. 2018, 69, 1235–1245. [Google Scholar] [CrossRef]
- Howe, G.A.; Major, I.T.; Koo, A.J. Modularity in Jasmonate Signaling for Multistress Resilience. Annu. Rev. Plant Biol. 2018, 69, 387–415. [Google Scholar] [CrossRef] [Green Version]
- Belete, T. Defense Mechanisms of Plants to Insect Pests: From Morphological to Biochemical A proach. Trends Tech. Sci. Res. 2018, 2, 555584. [Google Scholar] [CrossRef]
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of plant defense against insect herbivores. Plant Signal. Behav. 2012, 7, 1306–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.P.; Shahi, P.; Gase, K.; Baldwin, I.T. Herbivory-induced changes in the small-RNA transcriptome and phytohormone signaling in Nicotiana attenuata. Proc. Natl. Acad. Sci. USA 2008, 105, 4559–4564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, F.; Bodenhausen, N.; Lassueur, S.; Masclaux, F.G.; Reymond, P. Differential Contribution of Transcription Factors to Arabidopsis thaliana Defense Against Spodoptera littoralis. Front. Plant Sci. 2013, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.Z.; Chen, J.Y.; Xiao, H.J.; Xiao, Y.T.; Wu, J.; Wu, J.X.; Zhou, J.J.; Zhang, Y.J.; Guo, Y.Y. Dynamic transcriptome analysis and volatile profiling of Gossypium hirsutum in response to the cotton bollworm Helicoverpa armigera. Sci. Rep. 2015, 5, 11867. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.N.; Tang, L.; Hou, Y.; Wang, P.; Yang, H.; Wei, C.L. Differential transcriptome analysis of leaves of tea plant (Camellia sinensis) provides comprehensive insights into the defense responses to Ectropis oblique attack using RNA-Seq. Funct. Integr. Genom. 2016, 16, 383–398. [Google Scholar] [CrossRef]
- Ehlting, J.; Chowrira, S.G.; Mattheus, N.; Aeschliman, D.S.; Arimura, G.; Bohlmann, J. Comparative transcriptome analysis of Arabidopsis thaliana infested by diamond back moth (Plutella xylostella) larvae reveals signatures of stress response, secondary metabolism, and signalling. BMC Genom. 2008, 9, 154. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Ye, M.; Kuai, P.; Ye, M.; Erb, M.; Lou, Y. OsLRR-RLK1, an early responsive leucine-rich repeat receptor-like kinase, initiates rice defense responses against a chewing herbivore. New Phytol. 2018, 219, 1097–1111. [Google Scholar] [CrossRef] [Green Version]
- Hematy, K.; Sado, P.E.; Van Tuinen, A.; Rochange, S.; Desnos, T.; Balzergue, S.; Pelletier, S.; Renou, J.P.; Hofte, H. A receptor-like kinase mediates the response of Arabidopsis cells to the inhibition of cellulose synthesis. Curr. Biol. 2007, 17, 922–931. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Lin, W.; Zhou, X.; Guo, J.; Dang, X.; Li, B.; Lin, D.; Yang, Z. Mechano-transduction via the pectin-FERONIA complex activates ROP6 GTPase signaling in Arabidopsis pavement cell morphogenesis. Curr. Biol. 2022, 32, 508–517. [Google Scholar] [CrossRef]
- Feng, W.; Kita, D.; Peaucelle, A.; Cartwright, H.N.; Doan, V.; Duan, Q.; Liu, M.C.; Maman, J.; Steinhorst, L.; Schmitz-Thom, I.; et al. The FERONIA Receptor Kinase Maintains Cell-Wall Integrity during Salt Stress through Ca2+ Signaling. Curr. Biol. 2018, 28, 666–675. [Google Scholar] [CrossRef]
- Yan, C.; Fan, M.; Yang, M.; Zhao, J.; Zhang, W.; Su, Y.; Xiao, L.; Deng, H.; Xie, D. Injury Activates Ca2+/Calmodulin-Dependent Phosphorylation of JAV1-JAZ8-WRKY51 Complex for Jasmonate Biosynthesis. Mol. Cell 2018, 70, 136–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Zhang, W.; Guo, Y. Arabidopsis SOS3 plays an important role in salt tolerance by mediating calcium-dependent microfilament reorganization. Plant Cell Rep. 2013, 32, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Yang, D.; Ha, Y.; Lee, J.Y.; Kim, J.Y.; Oh, M.H.; Nam, K.H. Open stomata 1 exhibits dual serine/threonine and tyrosine kinase activity in regulating abscisic acid signaling. J. Exp. Bot. 2021, 72, 5494–5507. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Fischer, C.; Meldau, S.; Seebald, E.; Oelmuller, R.; Baldwin, I.T. Lipase activity in insect oral secretions mediates defense responses in Arabidopsis. Plant Physiol. 2011, 156, 1520–1534. [Google Scholar] [CrossRef] [Green Version]
- Glauser, G.; Dubugnon, L.; Mousavi, S.A.; Rudaz, S.; Wolfender, J.L.; Farmer, E.E. Velocity estimates for signal propagation leading to systemic jasmonic acid accumulation in wounded Arabidopsis. J. Biol. Chem. 2009, 284, 34506–34513. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, F.; Fernandez-Calvo, P.; Zander, M.; Diez-Diaz, M.; Fonseca, S.; Glauser, G.; Lewsey, M.G.; Ecker, J.R.; Solano, R.; Reymond, P. Arabidopsis basic helix-loop-helix transcription factors MYC2, MYC3, and MYC4 regulate glucosinolate biosynthesis, insect performance, and feeding behavior. Plant Cell 2013, 25, 3117–3132. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Huang, H.; Wang, J.; Liu, B.; Qi, T.; Xie, D. MYC5 is Involved in Jasmonate-Regulated Plant Growth, Leaf Senescence and Defense Responses. Plant Cell Physiol. 2017, 58, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Martin, C. Multifunctionality and diversity within the plant MYB-gene family. Plant Mol. Biol. 1999, 41, 577–585. [Google Scholar] [CrossRef]
- Feng, G.; Burleigh, J.G.; Braun, E.L.; Mei, W.; Barbazuk, W.B. Evolution of the 3R-MYB Gene Family in Plants. Genome Biol. Evol. 2017, 9, 1013–1029. [Google Scholar] [CrossRef] [Green Version]
- Dudareva, N.; Klempien, A.; Muhlemann, J.K.; Kaplan, I. Biosynthesis, function and metabolic engineering of plant volatile organic compounds. New Phytol. 2013, 198, 16–32. [Google Scholar] [CrossRef]
- Morawo, T.; Fadamiro, H. The role of herbivore- and plant-related experiences in intraspecific host preference of a relatively specialized parasitoid. Insect Sci. 2019, 26, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Dubey, N.K.; Goel, R.; Ranjan, A.; Idris, A.; Singh, S.K.; Bag, S.K.; Chandrashekar, K.; Pandey, K.D.; Singh, P.K.; Sawant, S.V. Comparative transcriptome analysis of Gossypium hirsutum L. in response to sap sucking insects: Aphid and whitefly. BMC Genom. 2013, 14, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Gene | Homolog Gene Name | EL_3h v CK L2fc | EL_6h v CK L2fc |
|---|---|---|---|---|
| Perception | Cluster-8107.7426 | LRR-RK | 2.099 | 2.368 |
| Cluster-8107.12313 | LRR-RK | 1.466 | - | |
| Cluster-8107.7028 | LRR-RK | 2.315 | 1.318 | |
| Cluster-6414.0 | THE1 | 2.731 | - | |
| Cluster-8107.18497 | FER | 2.476 | 1.555 | |
| Cluster-8107.8882 | RALFL33 | 2.274 | 1.355 | |
| Ca2+, ROS, MAPK signaling | Cluster-8107.14454 | CML30 | 2.096 | 1.488 |
| Cluster-8107.8609 | SOS3 | 1.700 | 2.016 | |
| Cluster-8107.24508 | PLP1 | 6.244 | 5.214 | |
| Cluster-8107.4341 | CDPK6 | 1.449 | 1.116 | |
| Cluster-8107.4806 | CPK15 | 2.759 | 2.193 | |
| Cluster-8107.23569 | RBOHD | 1.905 | 1.409 | |
| Cluster-8107.2579 | RBOH F | 3.997 | 2.992 | |
| Cluster-8107.12324 | MPK20 | 1.93 | 1.148 | |
| Cluster-8107.2274 | MAPKKK18 | 5.565 | 3.613 | |
| JA pathway | Cluster-8107.2271 | JAZ10 | 7.087 | 5.944 |
| Cluster-8107.14010 | JAZ3 | 3.977 | 3.320 | |
| Cluster-8107.11608 | JAZ1 | 3.341 | 2.389 | |
| Cluster-8107.12593 | JAR1 | 1.751 | - | |
| Cluster-8107.12373 | LOX2 | 3.154 | 2.330 | |
| Cluster-8107.7268 | AOS | 3.241 | 2.330 | |
| Cluster-8107.10842 | AOC | 5.685 | 4.150 | |
| Cluster-8107.11076 | OPR3 | 2.332 | 1.524 | |
| Cluster-8107.13500 | ACX1 | 2.330 | 1.302 | |
| Transcription factors | Cluster-8107.1997 | MYC3 | 6.610 | 6.012 |
| Cluster-8107.13830 | MYC4 | 2.550 | 2.057 | |
| Cluster-8107.13733 | MYC2 | 1.678 | 1.523 | |
| Cluster-8107.24465 | WRKY51 | 2.816 | - | |
| Cluster-8107.19064 | WRKY19 | 2.051 | 1.200 | |
| Cluster-8107.22388 | TTR1 | 2.019 | - | |
| Cluster-8107.15321 | WRKY3 | 3.348 | 2.428 | |
| Cluster-8107.20775 | WRKY40 | 2.107 | - | |
| Cluster-8107.9774 | MYB73 | 2.336 | - | |
| Cluster-8107.1983 | MYB14 | 9.886 | 9.061 | |
| Cluster-8107.1908 | MYB105 | 5.440 | 4.685 | |
| Cluster-8107.25130 | MYB62 | 4.537 | 3.073 | |
| Cluster-8107.27085 | MYB66 | 4.582 | - | |
| ROS | Cluster-8107.2992 | RAP2.11 | - | 2.033 |
| Cluster-8107.16572 | OST1 | 2.942 | 1.892 | |
| Cluster-8107.18090 | ARGAH2 | 5.257 | 4.760 | |
| Cluster-8107.19259 | UGT73B5 | 1.505 | - | |
| Cluster-8107.1416 | ABCG39 | 2.261 | 1.357 | |
| Cluster-8107.19231 | XF1 | 1.336 | - | |
| Cluster-8107.12560 | NRAMP2 | 1.020 | 1.079 | |
| Cluster-8107.11277 | APX1 | 1.142 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zhu, X.; Zhang, Y.; Wu, Z.; Fan, S.; Zhang, L. Comparative Transcriptome Analysis Identifies Key Defense Genes and Mechanisms in Mulberry (Morus alba) Leaves against Silkworms (Bombyx mori). Int. J. Mol. Sci. 2022, 23, 13519. https://doi.org/10.3390/ijms232113519
Zhang X, Zhu X, Zhang Y, Wu Z, Fan S, Zhang L. Comparative Transcriptome Analysis Identifies Key Defense Genes and Mechanisms in Mulberry (Morus alba) Leaves against Silkworms (Bombyx mori). International Journal of Molecular Sciences. 2022; 23(21):13519. https://doi.org/10.3390/ijms232113519
Chicago/Turabian StyleZhang, Xuejie, Xinxin Zhu, Yuqian Zhang, Zhicheng Wu, Shoujin Fan, and Luoyan Zhang. 2022. "Comparative Transcriptome Analysis Identifies Key Defense Genes and Mechanisms in Mulberry (Morus alba) Leaves against Silkworms (Bombyx mori)" International Journal of Molecular Sciences 23, no. 21: 13519. https://doi.org/10.3390/ijms232113519