
Spatial Memory Training Counteracts Hippocampal GIRK Channel Decrease in the Transgenic APPSw,Ind J9 Alzheimer’s Disease Mouse Model
 , , and
, , and
Abstract
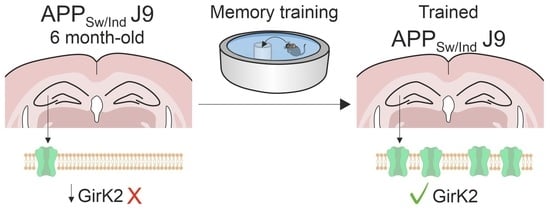
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. APPSw,Ind J9 Transgenic Mice
4.2. Immunohistochemical Staining
4.3. Western Blot
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-β |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| GIRK | G-protein-gated potassium channels |
| LTD | Long term depression |
| LTP | Long term potentiation |
| MWM | Morris water maze |
| RSG7 | G-protein signaling 7 |
| WT | Wild type |
References
- Gauthier, S.; Rosa-Neto, P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia; Alzheimer’s Disease International: London, UK, 2021. [Google Scholar]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Chin, J.; Palop, J.J.; Puoliväli, J.; Massaro, C.; Bien-Ly, N.; Gerstein, H.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2005, 25, 9694–9703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palop, J.J.; Jones, B.; Kekonius, L.; Chin, J.; Yu, G.Q.; Raber, J.; Masliah, E.; Mucke, L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc. Natl. Acad. Sci. USA 2003, 100, 9572–9577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.A.; Parra-Damas, A.; Enriquez-Barreto, L. Gene expression parallels synaptic excitability and plasticity changes in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra-Damas, A.; Valero, J.; Chen, M.; España, J.; Martín, E.; Ferrer, I.; Rodríguez-Alvarez, J.; Saura, C.A. Crtc1 activates a transcriptional program deregulated at early Alzheimer’s disease-related stages. J. Neurosci. 2014, 34, 5776–5787. [Google Scholar] [CrossRef] [Green Version]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [Green Version]
- Hector, A.; Brouillette, J. Hyperactivity Induced by Soluble Amyloid-β Oligomers in the Early Stages of Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 600084. [Google Scholar] [CrossRef]
- Dascal, N. Signalling via the G protein-activated K+ channels. Cell. Signal. 1997, 9, 551–573. [Google Scholar] [CrossRef]
- Djebari, S.; Iborra-Lázaro, G.; Temprano-Carazo, S.; Sánchez-Rodríguez, I.; Nava-Mesa, M.O.; Múnera, A.; Gruart, A.; Delgado-García, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. G-Protein-Gated Inwardly Rectifying Potassium (Kir3/GIRK) Channels Govern Synaptic Plasticity That Supports Hippocampal-Dependent Cognitive Functions in Male Mice. J. Neurosci. 2021, 41, 7086–7102. [Google Scholar] [CrossRef]
- Jeremic, D.; Sanchez-Rodriguez, I.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Therapeutic potential of targeting G protein-gated inwardly rectifying potassium (GIRK) channels in the central nervous system. Pharmacol. Ther. 2021, 223, 107808. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Slesinger, P.A. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010, 11, 301–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slesinger, P.A.; Wickman, K. Structure to Function of G Protein-Gated Inwardly Rectifying (GIRK) Channels.; Slesinger, P.A., Wickman, K., Eds.; Academic Press: Amsterdam, The Netherlands, 2015; p. 378. [Google Scholar]
- Karschin, C.; Dissmann, E.; Stühmer, W.; Karschin, A. IRK(1-3) and GIRK(1-4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J. Neurosci. 1996, 16, 3559–3570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pravetoni, M.; Wickman, K. Behavioral characterization of mice lacking GIRK/Kir3 channel subunits. Genes Brain Behav. 2008, 7, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Ostrovskaya, O.I.; Orlandi, C.; Fajardo-Serrano, A.; Young, S.M., Jr.; Lujan, R.; Martemyanov, K.A. Inhibitory Signaling to Ion Channels in Hippocampal Neurons Is Differentially Regulated by Alternative Macromolecular Complexes of RGS7. J. Neurosci. 2018, 38, 10002–10015. [Google Scholar] [CrossRef] [Green Version]
- Ostrovskaya, O.; Xie, K.; Masuho, I.; Fajardo-Serrano, A.; Lujan, R.; Wickman, K.; Martemyanov, K.A. RGS7/Gβ5/R7BP complex regulates synaptic plasticity and memory by modulating hippocampal GABABR-GIRK signaling. Elife 2014, 3, e02053. [Google Scholar] [CrossRef]
- Nava-Mesa, M.O.; Jiménez-Díaz, L.; Yajeya, J.; Navarro-Lopez, J.D. Amyloid-β induces synaptic dysfunction through G protein-gated inwardly rectifying potassium channels in the fimbria-CA3 hippocampal synapse. Front. Cell. Neurosci. 2013, 7, 117. [Google Scholar] [CrossRef] [Green Version]
- Mayordomo-Cava, J.; Yajeya, J.; Navarro-López, J.D.; Jiménez-Díaz, L. Amyloid-β(25-35) Modulates the Expression of GirK and KCNQ Channel Genes in the Hippocampus. PLoS ONE 2015, 10, e0134385. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rodríguez, I.; Gruart, A.; Delgado-García, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. Role of GirK Channels in Long-Term Potentiation of Synaptic Inhibition in an In Vivo Mouse Model of Early Amyloid-β Pathology. Int. J. Mol. Sci. 2019, 20, 1168. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rodríguez, I.; Temprano-Carazo, S.; Nájera, A.; Djebari, S.; Yajeya, J.; Gruart, A.; Delgado-García, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. Activation of G-protein-gated inwardly rectifying potassium (Kir3/GirK) channels rescues hippocampal functions in a mouse model of early amyloid-β pathology. Sci. Rep. 2017, 7, 14658. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, I.; Djebari, S.; Temprano-Carazo, S.; Vega-Avelaira, D.; Jiménez-Herrera, R.; Iborra-Lázaro, G.; Yajeya, J.; Jiménez-Díaz, L.; Navarro-López, J.D. Hippocampal long-term synaptic depression and memory deficits induced in early amyloidopathy are prevented by enhancing G-protein-gated inwardly rectifying potassium channel activity. J. Neurochem. 2019, 153, 362–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaro-Ruiz, R.; Martín-Belmonte, A.; Aguado, C.; Hernández, F.; Moreno-Martínez, A.E.; Ávila, J.; Luján, R. The Expression and Localisation of G-Protein-Coupled Inwardly Rectifying Potassium (GIRK) Channels Is Differentially Altered in the Hippocampus of Two Mouse Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11106. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.P.; Krohn, M.; Pahnke, J. Early Cognitive Training Rescues Remote Spatial Memory but Reduces Cognitive Flexibility in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2020, 75, 1301–1317. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Coria, H.; Yeung, S.T.; Ager, R.R.; Rodriguez-Ortiz, C.J.; Baglietto-Vargas, D.; LaFerla, F.M. Repeated cognitive stimulation alleviates memory impairments in an Alzheimer’s disease mouse model. Brain Res. Bull. 2015, 117, 10–15. [Google Scholar] [CrossRef]
- Oveisgharan, S.; Wilson, R.S.; Yu, L.; Schneider, J.A.; Bennett, D.A. Association of Early-Life Cognitive Enrichment With Alzheimer Disease Pathological Changes and Cognitive Decline. JAMA Neurol. 2020, 77, 1217–1224. [Google Scholar] [CrossRef]
- Hall, C.B.; Lipton, R.B.; Sliwinski, M.; Katz, M.J.; Derby, C.A.; Verghese, J. Cognitive activities delay onset of memory decline in persons who develop dementia. Neurology 2009, 73, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Bao, J.; Liu, W.; Gong, X.; Liang, Z.; Li, W.; Wu, M.; Xiao, Y.; Sun, B.; Wang, X.; et al. Spatial Training Attenuates Long-Term Alzheimer’s Disease-Related Pathogenic Processes in APP/PS1 Mice. J. Alzheimer’s Dis. 2022, 85, 1453–1466. [Google Scholar] [CrossRef]
- Mayordomo-Cava, J.; Iborra-Lázaro, G.; Djebari, S.; Temprano-Carazo, S.; Sánchez-Rodríguez, I.; Jeremic, D.; Gruart, A.; Delgado-García, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. Impairments of Synaptic Plasticity Induction Threshold and Network Oscillatory Activity in the Hippocampus Underlie Memory Deficits in a Non-Transgenic Mouse Model of Amyloidosis. Biology 2020, 9, 175. [Google Scholar] [CrossRef]
- Luján, R.; Marron Fernandez de Velasco, E.; Aguado, C.; Wickman, K. New insights into the therapeutic potential of Girk channels. Trends Neurosci. 2014, 37, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.; Grigoryan, G.; Guy-David, L.; Tsoory, M.M.; Chen, A.; Reuveny, E. Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 2642–2647. [Google Scholar] [CrossRef]
- Moncaster, J.A.; Pineda, R.; Moir, R.D.; Lu, S.; Burton, M.A.; Ghosh, J.G.; Ericsson, M.; Soscia, S.J.; Mocofanescu, A.; Folkerth, R.D.; et al. Alzheimer’s disease amyloid-beta links lens and brain pathology in Down syndrome. PLoS ONE 2010, 5, e10659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lott, I.T.; Head, E. Alzheimer disease and Down syndrome: Factors in pathogenesis. Neurobiol. Aging 2005, 26, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [Green Version]
- Akyuz, E.; Villa, C.; Beker, M.; Elibol, B. Unraveling the Role of Inwardly Rectifying Potassium Channels in the Hippocampus of an Aβ((1-42))-Infused Rat Model of Alzheimer’s Disease. Biomedicines 2020, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Alacid, L.; Watanabe, M.; Molnár, E.; Wickman, K.; Luján, R. Developmental regulation of G protein-gated inwardly-rectifying K+ (GIRK/Kir3) channel subunits in the brain. Eur J. Neurosci. 2011, 34, 1724–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disterhoft, J.F.; Wu, W.W.; Ohno, M. Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer’s disease. Ageing Res. Rev. 2004, 3, 383–406. [Google Scholar] [CrossRef]
- Barnes, C.A. Long-term potentiation and the ageing brain. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.C.; Norris, C.M. Age-associated changes in Ca(2+)-dependent processes: Relation to hippocampal synaptic plasticity. Hippocampus 1997, 7, 602–612. [Google Scholar] [CrossRef]
- Cohen, S.J.; Stackman, R.W., Jr. Assessing rodent hippocampal involvement in the novel object recognition task. A review. Behav. Brain Res. 2015, 285, 105–117. [Google Scholar] [CrossRef]
- Clarke, J.R.; Cammarota, M.; Gruart, A.; Izquierdo, I.; Delgado-García, J.M. Plastic modifications induced by object recognition memory processing. Proc. Natl. Acad. Sci. USA 2010, 107, 2652–2657. [Google Scholar] [CrossRef]
- Warburton, E.C.; Brown, M.W. Findings from animals concerning when interactions between perirhinal cortex, hippocampus and medial prefrontal cortex are necessary for recognition memory. Neuropsychologia 2010, 48, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Gold, G.; von Gunten, A.; Hof, P.R.; Bouras, C. Pathological substrates of cognitive decline in Alzheimer’s disease. Front. Neurol. Neurosci. 2009, 24, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.H.; Hof, P.R. Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer’s disease. Prog. Brain Res. 2002, 136, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forloni, G.; Lucca, E.; Angeretti, N.; Della Torre, P.; Salmona, M. Amidation of beta-amyloid peptide strongly reduced the amyloidogenic activity without alteration of the neurotoxicity. J. Neurochem. 1997, 69, 2048–2054. [Google Scholar] [CrossRef]
- West, M.J.; Coleman, P.D.; Flood, D.G.; Troncoso, J.C. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 1994, 344, 769–772. [Google Scholar] [CrossRef]
- Balducci, C.; Forloni, G. APP transgenic mice: Their use and limitations. Neuromolecular Med. 2011, 13, 117–137. [Google Scholar] [CrossRef]
- Double, K.L.; Reyes, S.; Werry, E.L.; Halliday, G.M. Selective cell death in neurodegeneration: Why are some neurons spared in vulnerable regions? Prog. Neurobiol. 2010, 92, 316–329. [Google Scholar] [CrossRef]
- Marron Fernandez de Velasco, E.; Zhang, L.; Vo, B.N.; Tipps, M.; Farris, S.; Xia, Z.; Anderson, A.; Carlblom, N.; Weaver, C.D.; Dudek, S.M.; et al. GIRK2 splice variants and neuronal G protein-gated K(+) channels: Implications for channel function and behavior. Sci. Rep. 2017, 7, 1639. [Google Scholar] [CrossRef]
- Hall, A.M.; Throesch, B.T.; Buckingham, S.C.; Markwardt, S.J.; Peng, Y.; Wang, Q.; Hoffman, D.A.; Roberson, E.D. Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J. Neurosci. 2015, 35, 6221–6230. [Google Scholar] [CrossRef] [Green Version]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fülöp, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkänen, A.; et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toniolo, S.; Sen, A.; Husain, M. Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9318. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Konnerth, A. Neuronal hyperactivity--A key defect in Alzheimer’s disease? Bioessays 2015, 37, 624–632. [Google Scholar] [CrossRef]
- Maestú, F.; de Haan, W.; Busche, M.A.; DeFelipe, J. Neuronal excitation/inhibition imbalance: Core element of a translational perspective on Alzheimer pathophysiology. Ageing Res. Rev. 2021, 69, 101372. [Google Scholar] [CrossRef]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What Happens with the Circuit in Alzheimer’s Disease in Mice and Humans? Annu Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzowski, J.F.; Setlow, B.; Wagner, E.K.; McGaugh, J.L. Experience-dependent gene expression in the rat hippocampus after spatial learning: A comparison of the immediate-early genes Arc, c-fos, and zif268. J. Neurosci. 2001, 21, 5089–5098. [Google Scholar] [CrossRef] [Green Version]
- España, J.; Giménez-Llort, L.; Valero, J.; Miñano, A.; Rábano, A.; Rodriguez-Alvarez, J.; LaFerla, F.M.; Saura, C.A. Intraneuronal beta-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer’s disease transgenic mice. Biol. Psychiatry 2010, 67, 513–521. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Reference | Supplier | Host | Dilution |
|---|---|---|---|---|
| GIRK1 | APC-005 | Alomone, Jerusalem, Israel | Rabbit | 1/500 |
| GIRK2 | APC-006 | Alomone, Jerusalem, Israel | Rabbit | 1/500 |
| GIRK3 | APC-038 | Alomone, Jerusalem, Israel | Rabbit | 1/500 |
| GIRK4 | APC-027 | Alomone, Jerusalem, Israel | Rabbit | 1/500 |
| RGS7 | SC-8139 | Santa Cruz Biotech, Dallas, TX, US | Goat | 1/500 |
| β-actin | AC-15 | Sigma Aldrich, St. Louis, MO, US | Mouse | 1:100000 |
| Rabbit IgG-HRP | 170-6515 | Bio-Rad, Hercules, CA, US | Goat | 1/3000 |
| Mouse IgG-HRP | 170-6516 | Bio-Rad, Hercules, CA, US | Goat | 1/3000 |
| Goat IgG-HRP | SC-2352 | Merck—Millipore, Burlington, MA, US | Monkey | 1/3000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temprano-Carazo, S.; Contreras, A.; Saura, C.A.; Navarro-López, J.D.; Jiménez-Díaz, L. Spatial Memory Training Counteracts Hippocampal GIRK Channel Decrease in the Transgenic APPSw,Ind J9 Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2022, 23, 13444. https://doi.org/10.3390/ijms232113444
Temprano-Carazo S, Contreras A, Saura CA, Navarro-López JD, Jiménez-Díaz L. Spatial Memory Training Counteracts Hippocampal GIRK Channel Decrease in the Transgenic APPSw,Ind J9 Alzheimer’s Disease Mouse Model. International Journal of Molecular Sciences. 2022; 23(21):13444. https://doi.org/10.3390/ijms232113444
Chicago/Turabian StyleTemprano-Carazo, Sara, Ana Contreras, Carlos A. Saura, Juan D. Navarro-López, and Lydia Jiménez-Díaz. 2022. "Spatial Memory Training Counteracts Hippocampal GIRK Channel Decrease in the Transgenic APPSw,Ind J9 Alzheimer’s Disease Mouse Model" International Journal of Molecular Sciences 23, no. 21: 13444. https://doi.org/10.3390/ijms232113444