
Oligomerization of Human Cystatin C—An Amyloidogenic Protein: An Analysis of Small Oligomeric Subspecies
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Behaviour of Human Cystatin C under Various pH Conditions
2.2. Characterization of Oligomeric Species Present in HCC Solution at pH 3
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Protein Oligomerization
4.3. Dynamic Light Scattering
4.4. Small-Angle X-ray Scattering
4.5. SAXS Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grubb, A.; Löfberg, H. Human Gamma-Trace, a Basic Microprotein: Amino Acid Sequence and Presence in the Adenohypophysis. Proc. Natl. Acad. Sci. USA 1982, 79, 3024–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzin, J.; Popovič, T.; Turk, V.; Borchart, U.; Machleidt, W. Human Cystatin, a New Protein Inhibitor of Cysteine Proteinases. Biochem. Biophys. Res. Commun. 1984, 118, 103–109. [Google Scholar] [CrossRef]
- Abrahamson, M.; Mason, R.W.; Hansson, H.; Buttle, D.J.; Grubb, A.; Ohlsson, K. Human Cystatin C. Role of the N -Terminal Segment in the Inhibition of Human Cysteine Proteinases and in Its Inactivation by Leucocyte Elastase. Biochem. J. 1991, 273, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamson, M.; Ritonja, A.; Brown, M.A.; Grubb, A.; Machleidt, W.; Barrett, A.J. Identification of the Probable Inhibitory Reactive Sites of the Cysteine Proteinase Inhibitors Human Cystatin C and Chicken Cystatin. J. Biol. Chem. 1987, 262, 9688–9694. [Google Scholar] [CrossRef]
- Janowski, R.; Kozak, M.; Jankowska, E.; Grzonka, Z.; Grubb, A.; Abrahamson, M.; Jaskolski, M. Human Cystatin C, an Amyloidogenic Protein, Dimerizes through Three-Dimensional Domain Swapping. Nat. Struct Biol. 2001, 8, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Kozak, M.; Jankowska, E.; Janowski, R.; Grzonka, Z.; Grubb, A.; Alvarez Fernandez, M.; Abrahamson, M.; Jaskolski, M. Expression of a Selenomethionyl Derivative and Preliminary Crystallographic Studies of Human Cystatin C. Acta Cryst. D Biol. Cryst. 1999, 55, 1939–1942. [Google Scholar] [CrossRef] [Green Version]
- Janowski, R.; Abrahamson, M.; Grubb, A.; Jaskolski, M. Domain Swapping in N-Truncated Human Cystatin C. J. Mol. Biol. 2004, 341, 151–160. [Google Scholar] [CrossRef]
- Kolodziejczyk, R.; Michalska, K.; Hernandez-Santoyo, A.; Wahlbom, M.; Grubb, A.; Jaskolski, M. Crystal Structure of Human Cystatin C Stabilized against Amyloid Formation. FEBS J. 2010, 277, 1726–1737. [Google Scholar] [CrossRef]
- Maszota-Zieleniak, M.; Jurczak, P.; Orlikowska, M.; Zhukov, I.; Borek, D.; Otwinowski, Z.; Skowron, P.; Pietralik, Z.; Kozak, M.; Szymańska, A.; et al. NMR and Crystallographic Structural Studies of the Extremely Stable Monomeric Variant of Human Cystatin C with Single Amino Acid Substitution. FEBS J. 2020, 287, 361–376. [Google Scholar] [CrossRef]
- Janowski, R.; Kozak, M.; Abrahamson, M.; Grubb, A.; Jaskolski, M. 3D Domain-Swapped Human Cystatin C with Amyloidlike Intermolecular Beta-Sheets. Proteins 2005, 61, 570–578. [Google Scholar] [CrossRef]
- Åkesson, A.; Lindström, V.; Nyman, U.; Jonsson, M.; Abrahamson, M.; Christensson, A.; Björk, J.; Grubb, A. Shrunken Pore Syndrome and Mortality: A Cohort Study of Patients with Measured GFR and Known Comorbidities. Scand. J. Clin. Lab. Investig. 2020, 80, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Grubb, A. Shrunken Pore Syndrome—A Common Kidney Disorder with High Mortality. Diagnosis, Prevalence, Pathophysiology and Treatment Options. Clin. Biochem. 2020, 83, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, J.; Jensson, O.; Frangione, B. Amyloid Fibrils in Hereditary Cerebral Hemorrhage with Amyloidosis of Icelandic Type Is a Variant of Gamma-Trace Basic Protein (Cystatin C). Proc. Natl. Acad. Sci. USA 1986, 83, 2974–2978. [Google Scholar] [CrossRef] [Green Version]
- Palsdottir, A.; Snorradottir, A.O.; Thorsteinsson, L. Hereditary Cystatin C Amyloid Angiopathy: Genetic, Clinical, and Pathological Aspects. Brain Pathol. 2006, 16, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Ólafsson, Í.; Thorsteinsson, L.; Jensson, Ó. The Molecular Pathology of Hereditary Cystatin C Amyloid Angiopathy Causing Brain Hemorrhage. Brain Pathol. 1996, 6, 121–126. [Google Scholar] [CrossRef]
- Graffagnino, C.; Herbstreith, M.H.; Schmechel, D.E.; Levy, E.; Roses, A.D.; Alberts, M.J. Cystatin C Mutation in an Elderly Man with Sporadic Amyloid Angiopathy and Intracerebral Hemorrhage. Stroke 1995, 26, 2190–2193. [Google Scholar] [CrossRef] [PubMed]
- Mathews, P.M.; Levy, E. Cystatin C in Aging and in Alzheimer’s Disease. Ageing Res. Rev. 2016, 32, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Östner, G.; Lindström, V.; Hjort Christensen, P.; Kozak, M.; Abrahamson, M.; Grubb, A. Stabilization, Characterization, and Selective Removal of Cystatin C Amyloid Oligomers. J. Biol. Chem. 2013, 288, 16438–16450. [Google Scholar] [CrossRef] [Green Version]
- Chrabąszczewska, M.; Sieradzan, A.K.; Rodziewicz-Motowidło, S.; Grubb, A.; Dobson, C.M.; Kumita, J.R.; Kozak, M. Structural Characterization of Covalently Stabilized Human Cystatin C Oligomers. Int. J. Mol. Sci. 2020, 21, 5860. [Google Scholar] [CrossRef]
- Chrabąszczewska, M.; Maszota-Zieleniak, M.; Pietralik, Z.; Taube, M.; Rodziewicz-Motowidło, S.; Szymańska, A.; Szutkowski, K.; Clemens, D.; Grubb, A.; Kozak, M. Cyclic Trimer of Human Cystatin C, an Amyloidogenic Protein—Molecular Dynamics and Experimental Studies. J. Appl. Phys. 2018, 123, 174701. [Google Scholar] [CrossRef]
- Da Vela, S.; Svergun, D.I. Methods, Development and Applications of Small-Angle X-Ray Scattering to Characterize Biological Macromolecules in Solution. Curr. Res. Struct. Biol. 2020, 2, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Jacques, D.A.; Trewhella, J. Small-Angle Scattering for Structural Biology-Expanding the Frontier While Avoiding the Pitfalls: Small-Angle Scattering for Structural Biology. Protein Sci. 2010, 19, 642–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, M.; Pietralik, Z.; Szymanska, A.; Szutkowski, K.; Clemens, D.; Grubb, A.; Kozak, M. The Domain Swapping of Human Cystatin C Induced by Synchrotron Radiation. Sci. Rep. 2019, 9, 8548. [Google Scholar] [CrossRef] [Green Version]
- Good, N.E.; Winget, G.D.; Winter, W.; Connolly, T.N.; Izawa, S.; Singh, R.M.M. Hydrogen Ion Buffers for Biological Research. Biochemistry 1966, 5, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Wahlbom, M.; Wang, X.; Lindström, V.; Carlemalm, E.; Jaskolski, M.; Grubb, A. Fibrillogenic Oligomers of Human Cystatin C Are Formed by Propagated Domain Swapping. J. Biol. Chem. 2007, 282, 18318–18326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlenfein, T.J.; Mehlhoff, J.D.; Murphy, R.M. Insights into the Mechanism of Cystatin C Oligomer and Amyloid Formation and Its Interaction with β-Amyloid. J. Biol. Chem. 2017, 292, 11485–11498. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, M.; Wang, X.; Rodziewicz-Motowidlo, S.; Janowski, R.; Lindström, V.; Önnerfjord, P.; Westermark, G.; Grzonka, Z.; Jaskolski, M.; Grubb, A. Prevention of Domain Swapping Inhibits Dimerization and Amyloid Fibril Formation of Cystatin C. J. Biol. Chem. 2004, 279, 24236–24245. [Google Scholar] [CrossRef] [Green Version]
- Dall, E.; Hollerweger, J.C.; Dahms, S.O.; Cui, H.; Häussermann, K.; Brandstetter, H. Structural and Functional Analysis of Cystatin E Reveals Enzymologically Relevant Dimer and Amyloid Fibril States. J. Biol. Chem. 2018, 293, 13151–13165. [Google Scholar] [CrossRef] [Green Version]
- Shingate, P.; Warwicker, J.; Sowdhamini, R. Energetic Calculations to Decipher PH-Dependent Oligomerization and Domain Swapping of Proteins. PLoS ONE 2015, 10, e0127716. [Google Scholar] [CrossRef] [Green Version]
- Baler, K.; Martin, O.A.; Carignano, M.A.; Ameer, G.A.; Vila, J.A.; Szleifer, I. Electrostatic Unfolding and Interactions of Albumin Driven by PH Changes: A Molecular Dynamics Study. J. Phys. Chem. B 2014, 118, 921–930. [Google Scholar] [CrossRef]
- Santos, J.; Iglesias, V.; Santos-Suárez, J.; Mangiagalli, M.; Brocca, S.; Pallarès, I.; Ventura, S. PH-Dependent Aggregation in Intrinsically Disordered Proteins Is Determined by Charge and Lipophilicity. Cells 2020, 9, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batoulis, H.; Schmidt, T.H.; Weber, P.; Schloetel, J.-G.; Kandt, C.; Lang, T. Concentration Dependent Ion-Protein Interaction Patterns Underlying Protein Oligomerization Behaviours. Sci. Rep. 2016, 6, 24131. [Google Scholar] [CrossRef] [Green Version]
- Samantray, S.; Olubiyi, O.O.; Strodel, B. The Influences of Sulphation, Salt Type, and Salt Concentration on the Structural Heterogeneity of Glycosaminoglycans. Int. J. Mol. Sci. 2021, 22, 11529. [Google Scholar] [CrossRef]
- Tovar-Anaya, D.O.; Vera-Robles, L.I.; Vieyra-Eusebio, M.T.; García-Gutiérrez, P.; Reyes-Espinosa, F.; Hernández-Arana, A.; Arroyo-Reyna, J.A.; Zubillaga, R.A. Stabilized Human Cystatin C Variant L47C/G69C Is a Better Reporter Than the Wild-Type Inhibitor for Characterizing the Thermodynamics of Binding to Cysteine Proteases. Protein J 2019, 38, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Girardot, R.; Viguier, G.; Pérez, J.; Ounsy, M. FOXTROT: A JAVA-Based Application to Reduce and Analyse SAXS and WAXS Piles of 2D Data at Synchrotron SOLEIL, Synchrotron Soleil, Saint-Aubin, France. In Proceedings of the canSAS-VIII, Tokai, Japan, 14–16 April 2015. [Google Scholar]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-Based System for Small-Angle Scattering Data Analysis. J. Appl. Cryst. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded Functionality and New Tools for Small-Angle Scattering Data Analysis. J. Appl. Cryst. 2021, 54, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL—A Program to Evaluate X-Ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Cryst. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Svergun, D.I. Global Rigid Body Modeling of Macromolecular Complexes against Small-Angle Scattering Data. Biophys. J. 2005, 89, 1237–1250. [Google Scholar] [CrossRef] [Green Version]
- Volkov, V.V.; Svergun, D.I. Uniqueness of Ab Initio Shape Determination in Small-Angle Scattering. J. Appl. Cryst. 2003, 36, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Franke, D.; Svergun, D.I. DAMMIF, a Program for Rapid Ab-Initio Shape Determination in Small-Angle Scattering. J. Appl. Cryst. 2009, 42, 342–346. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehnal, D.; Bittrich, S.; Deshpande, R.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucl. Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]








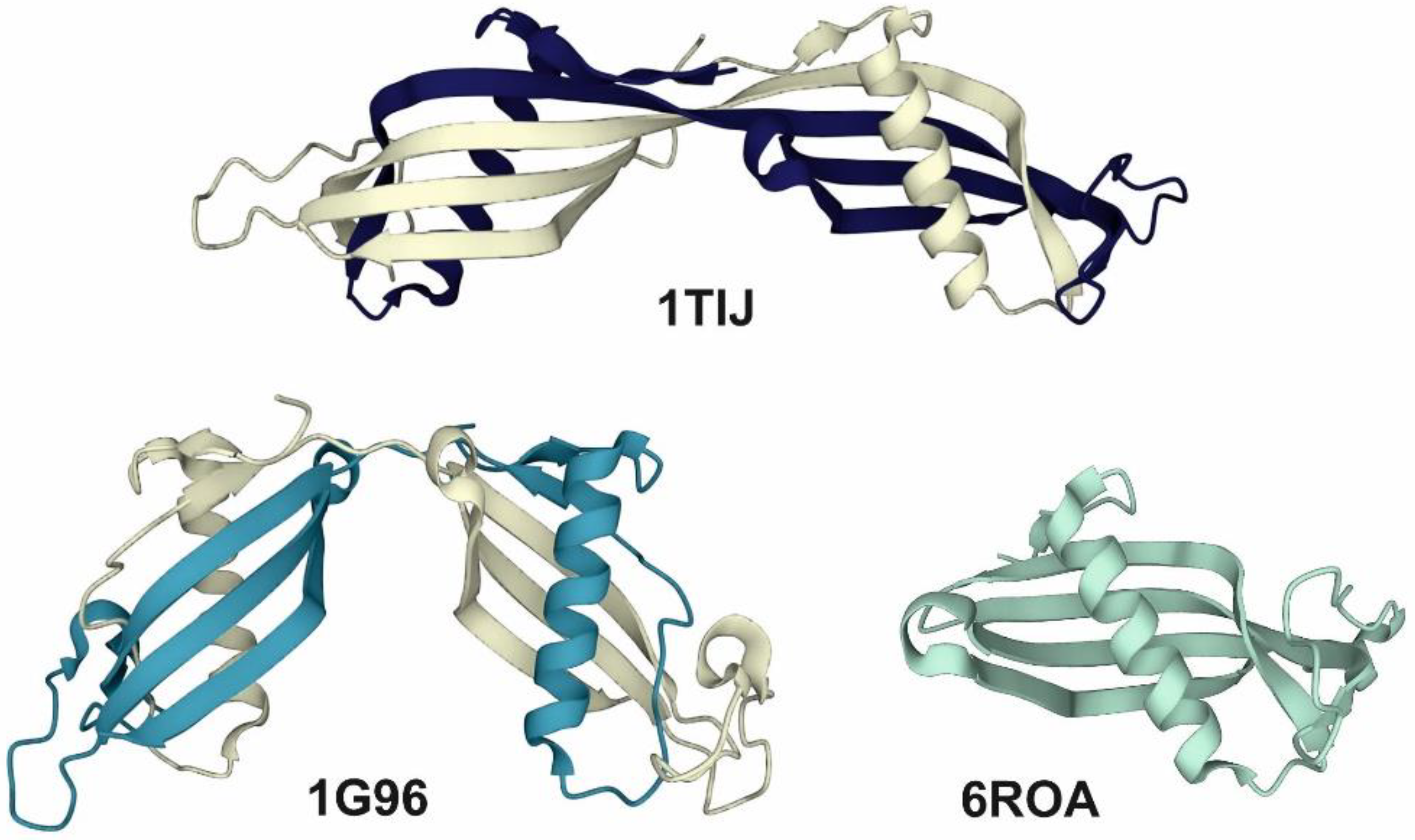{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
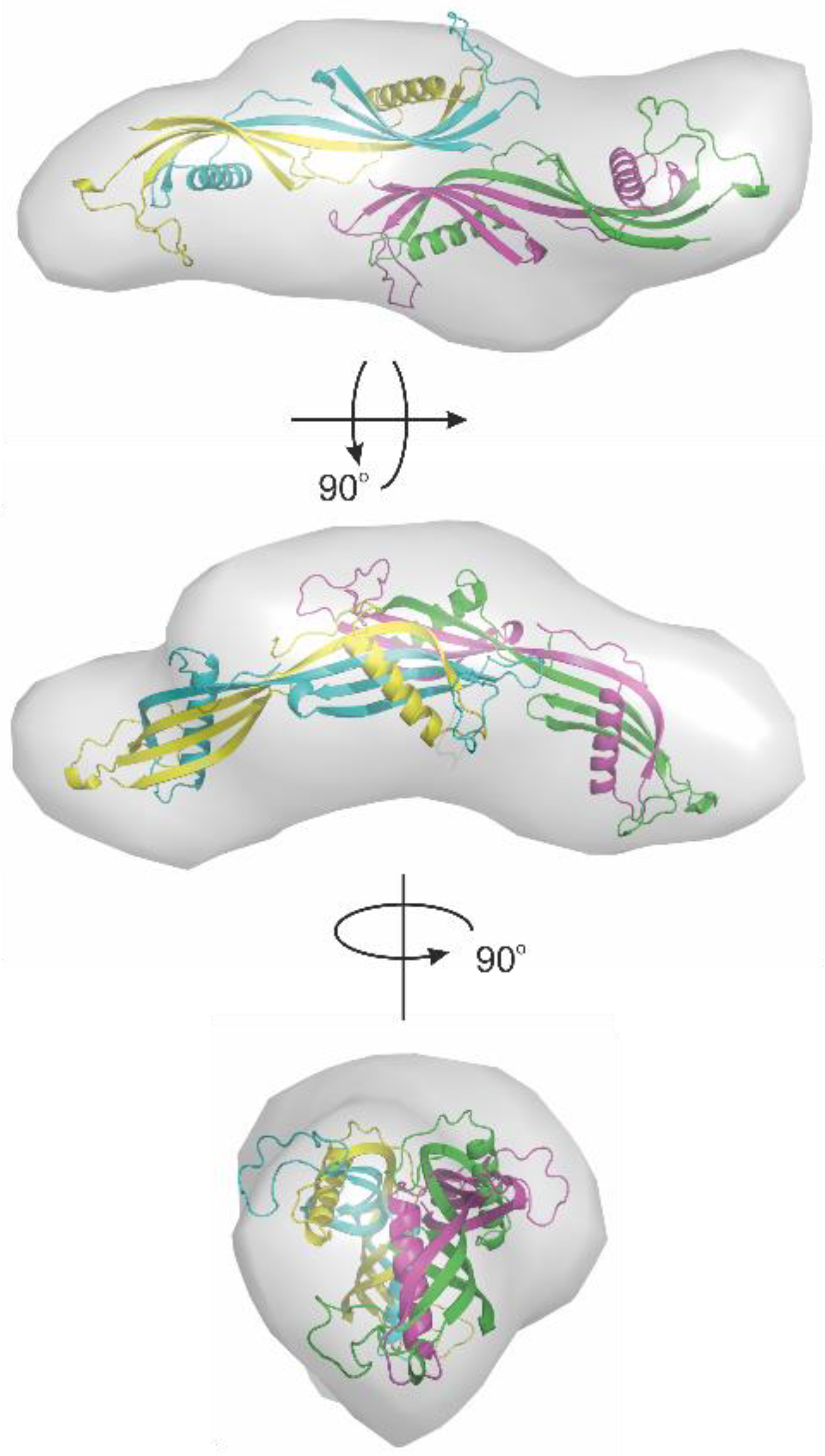{kind=link}
| Sample | RG [nm] | Dmax [nm] | RH [nm] |
|---|---|---|---|
| Glycine pH 2.2 | 3.91 ± 0.1 | 17.0 | 3.16 ± 0.1 |
| Citric acid pH 3 | 3.42 ± 0.1 | 14.0 | 3.21 ± 0.06 |
| Sodium acetate pH 4 | 2.64 ± 0.05 | 12.0 | 2.22 ± 0.06 |
| Sodium acetate pH 4 + GdmCl | 2.98 ± 0.08 | 12.5 | 2.15 ± 0.06 |
| Citric acid pH 5 | 2.41 ± 0.07 | 11.2 | 2.47 ± 0.15 |
| MES pH 6 | 1.55 ± 0.04 | 6.0 | 1.50 ± 0.05 |
| Citric acid pH 7 | 1.64 ± 0.05 | 6.7 | 1.51 ± 0.02 |
| aCSF pH 7.4 | 1.76 ± 0.03 | 8.0 | 1.63 ± 0.05 |
| PBS pH 7.4 | 1.63 ± 0.03 | 6.3 | 1.53 ± 0.05 |
| PBS pH 7.4 + GdmCl | 1.63 ± 0.12 | 6.6 | 1.47 ± 0.05 |
| Tricine pH 8.8 | 1.68 ± 0.07 | 6.2 | 1.48 ± 0.05 |
| Glycine pH 10 | 1.73 ± 0.05 | 7.8 | 1.57 ± 0.07 |
| Sample | Monomer | Dimer | Tetramer |
|---|---|---|---|
| pH 3 to pH 7.4 (PBS, First 10 min) | 0.07 | 0.00 | 0.92 |
| pH 3 to pH 7.4 (PBS, 20–1 h 20 min) | 0.00 | 1.00 | 0.00 |
| pH 3 to pH 7.4 (20 mM Tris, 50 mM NaCl) | 0.00 | 1.00 | - |
| pH 3 to pH 3 | 0.00 | 0.00 | 0.99 |
| Sample | Monomer | Dimer | Tetramer |
|---|---|---|---|
| Glycine pH 2.2 | 0.00 | 0.00 | 1.00 |
| Citric acid pH 3 | 0.00 | 0.00 | 1.00 |
| Sodium acetate pH 4 | 0.00 | 0.85 | 0.15 |
| Sodium acetate pH 4 + GdmCl | 0.00 | 0.70 | 0.30 |
| Citric acid pH 5 | 0.07 | 0.93 | - |
| MES pH 6 | 0.97 | 0.03 | - |
| Citric acid pH 7 | 0.84 | 0.16 | - |
| aCSF pH 7.4 | 0.83 | 0.17 | - |
| PBS pH 7.4 | 0.89 | 0.10 | - |
| PBS pH 7.4 + GdmCl | 0.93 | 0.00 | 0.07 |
| Tricine pH 8.8 | 0.87 | 0.13 | - |
| Glycine pH 10 | 0.85 | 0.15 | - |
| Buffer | Final Buffer Composition 1 | pH 2 |
|---|---|---|
| Glycine pH 2.2 | 50 mM Glycine, 100 mM NaCl | 2.2 |
| Citric acid pH 3 | 50 mM Citric acid, 100 mM NaCl | 3 |
| Sodium acetate pH 4 | 50 mM Sodium acetate, 100 mM NaCl | 4 |
| Sodium acetate pH 4 + GdmCl | 50 mM Sodium acetate, 100 mM NaCl, 0.2 M guanidinium chloride | 4 |
| Citric acid pH 5 | 50 mM Citric acid, 100 mM NaCl | 5 |
| MES pH 6 | 50 mM MES, 100 mM NaCl | 6 |
| Citric acid pH 7 | 50 mM Citric acid, 100 mM NaCl | 7 |
| aCSF pH 7.4 | 5.4 mM KCl, 5 mM Hepes, 1.8 mM CaCl2, 1 mM MgCl2, 135 mM NaCl | 7.4 |
| PBS pH 7.4 | 2.7 mM KCl, 1.8 mM KH2PO4, 0.9 mM Na3PO4, 137 mM NaCl 3 | 7.4 |
| PBS pH 7.4 + GdmCl | 2.7 mM KCl, 1.8 mM KH2PO4, 0.9 mM Na3PO4, 137 mM NaCl 3, 0.2 M guanidinium chloride | 7.4 |
| Tricine pH 8.8 | 50 mM Tricine, 100 mM NaCl | 8.8 |
| Glycine pH 10 | 50 mM Glycine, 100 mM NaCl | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojciechowska, D.; Taube, M.; Rucińska, K.; Maksim, J.; Kozak, M. Oligomerization of Human Cystatin C—An Amyloidogenic Protein: An Analysis of Small Oligomeric Subspecies. Int. J. Mol. Sci. 2022, 23, 13441. https://doi.org/10.3390/ijms232113441
Wojciechowska D, Taube M, Rucińska K, Maksim J, Kozak M. Oligomerization of Human Cystatin C—An Amyloidogenic Protein: An Analysis of Small Oligomeric Subspecies. International Journal of Molecular Sciences. 2022; 23(21):13441. https://doi.org/10.3390/ijms232113441
Chicago/Turabian StyleWojciechowska, Daria, Michał Taube, Karolina Rucińska, Joanna Maksim, and Maciej Kozak. 2022. "Oligomerization of Human Cystatin C—An Amyloidogenic Protein: An Analysis of Small Oligomeric Subspecies" International Journal of Molecular Sciences 23, no. 21: 13441. https://doi.org/10.3390/ijms232113441