
Molecular Mechanisms Linking Inflammation to Autoimmunity in Sjögren’s Syndrome: Identification of New Targets

Abstract
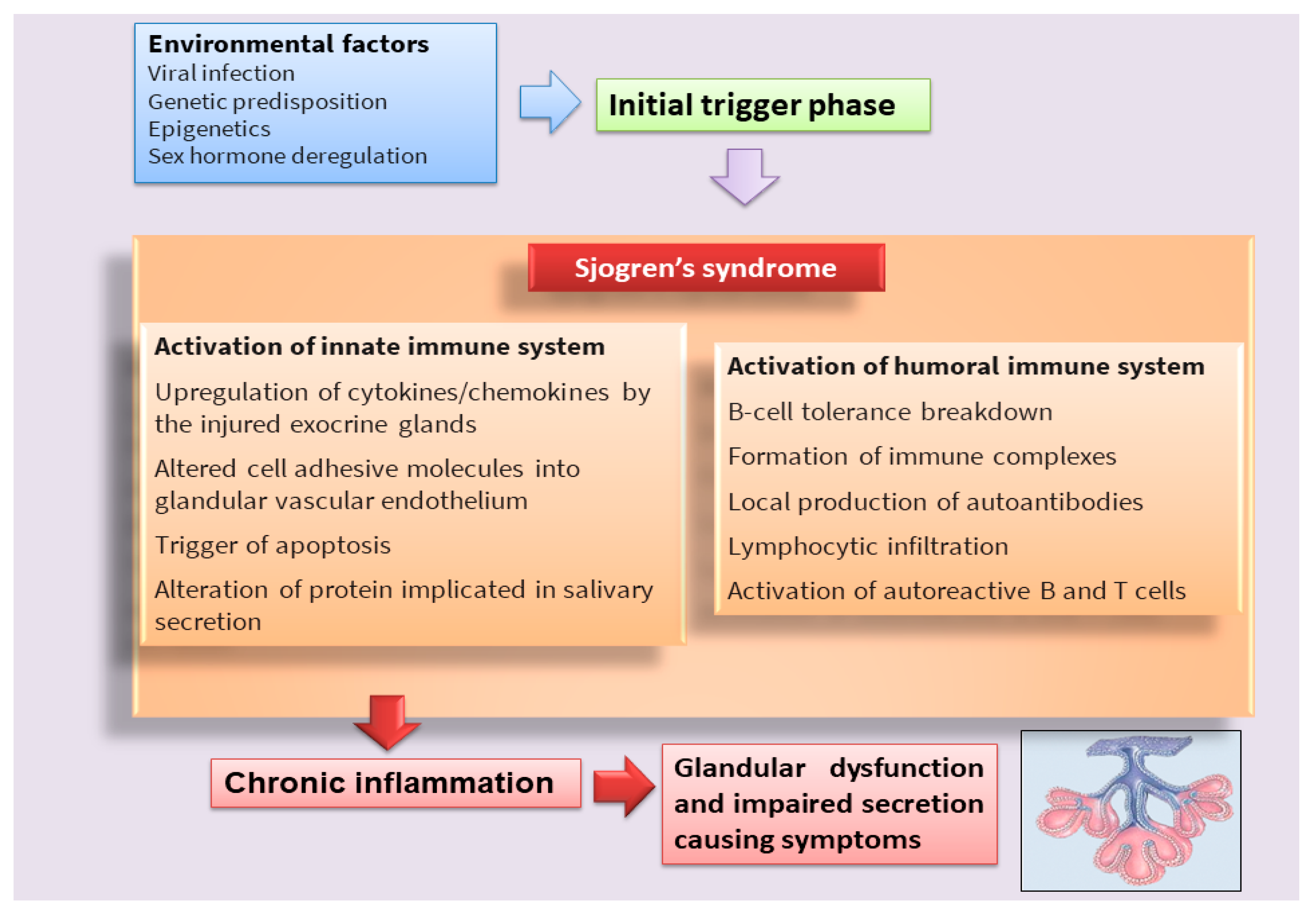
:1. Introduction
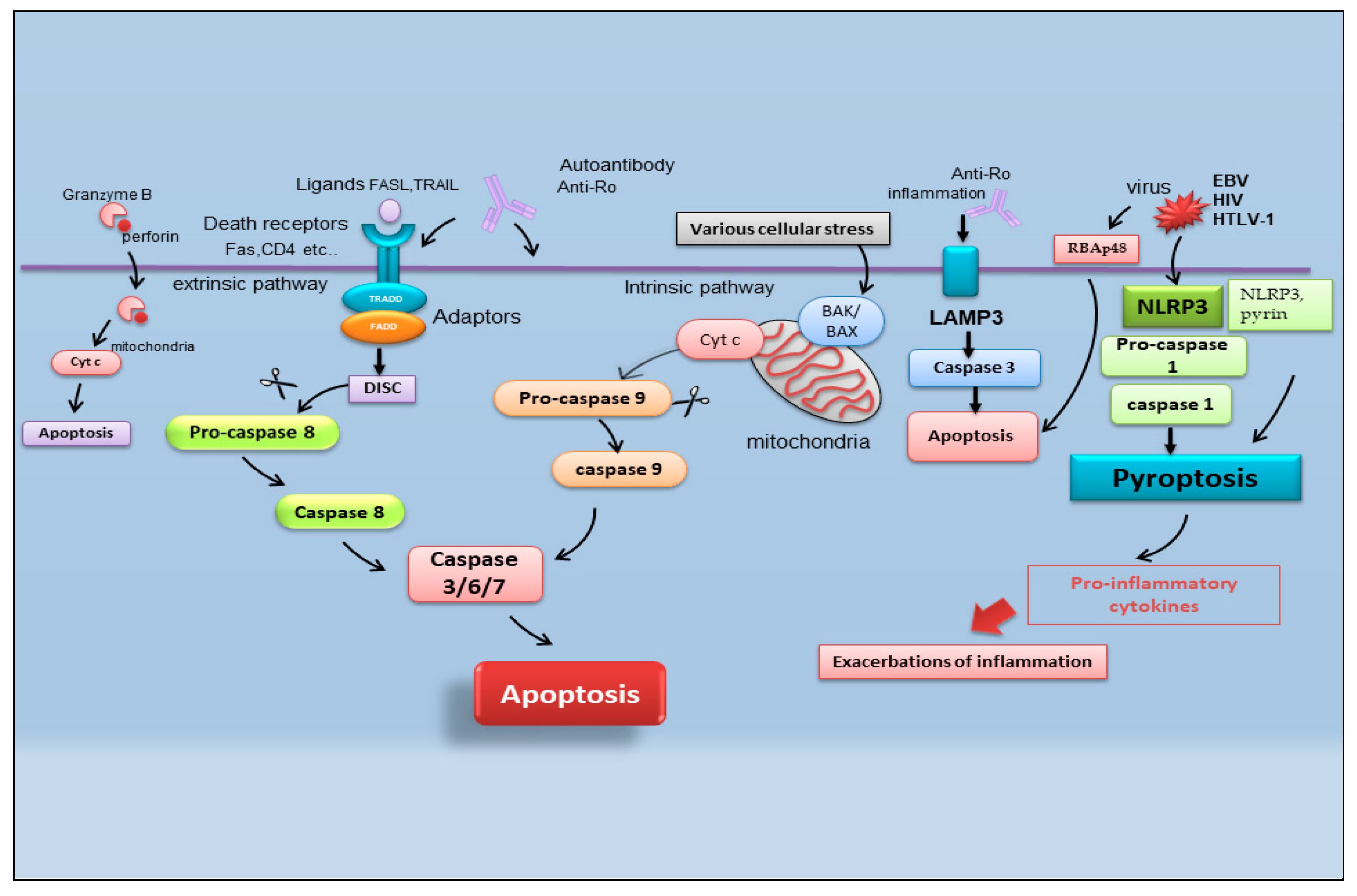
2. Recent Advances in Apoptosis in SS
2.1. Pyroptosis
2.2. Apoptosis and Viral Infection in SS
2.3. Lysosome-Associated Membrane Protein 3-Dependent Apoptosis
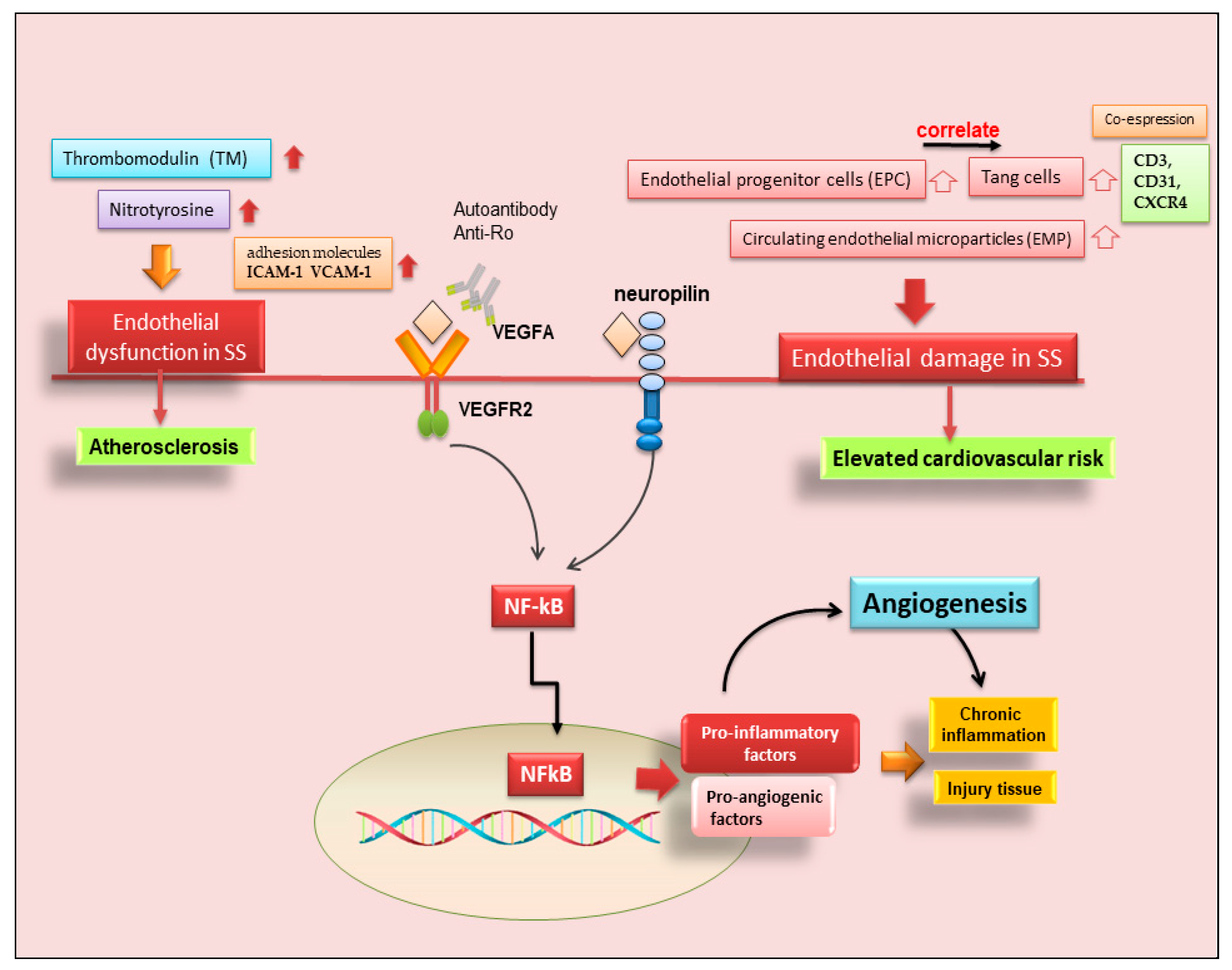
3. Angiogenesis in SS
3.1. Neo-Angiogenesis in pSS SGs
3.2. Endothelial T Cells in pSS
3.3. Possible Occurrence of Atherosclerosis in SS
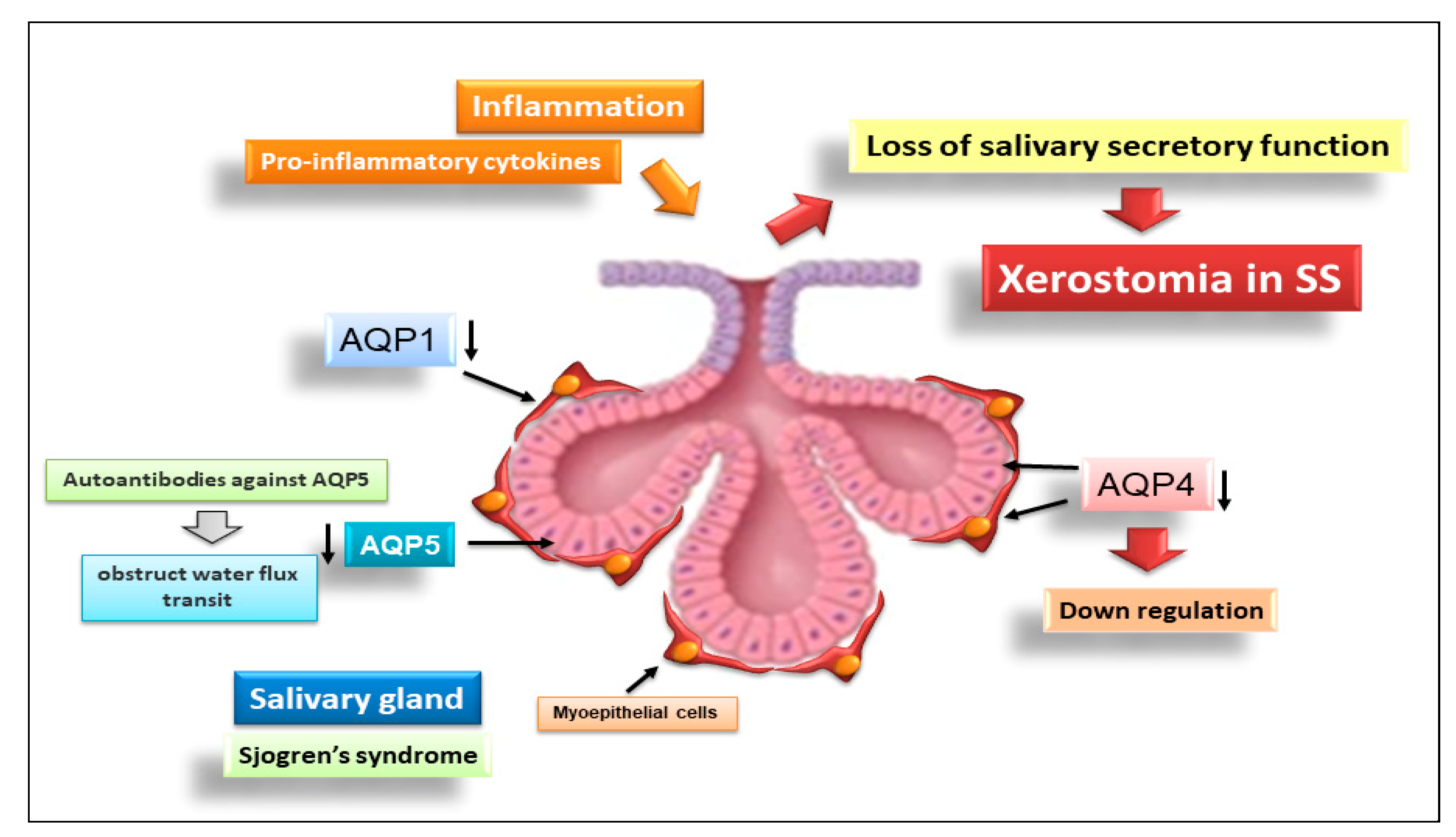
4. Novel Insights into the Role of Aquaporins in the Pathogenesis of SS
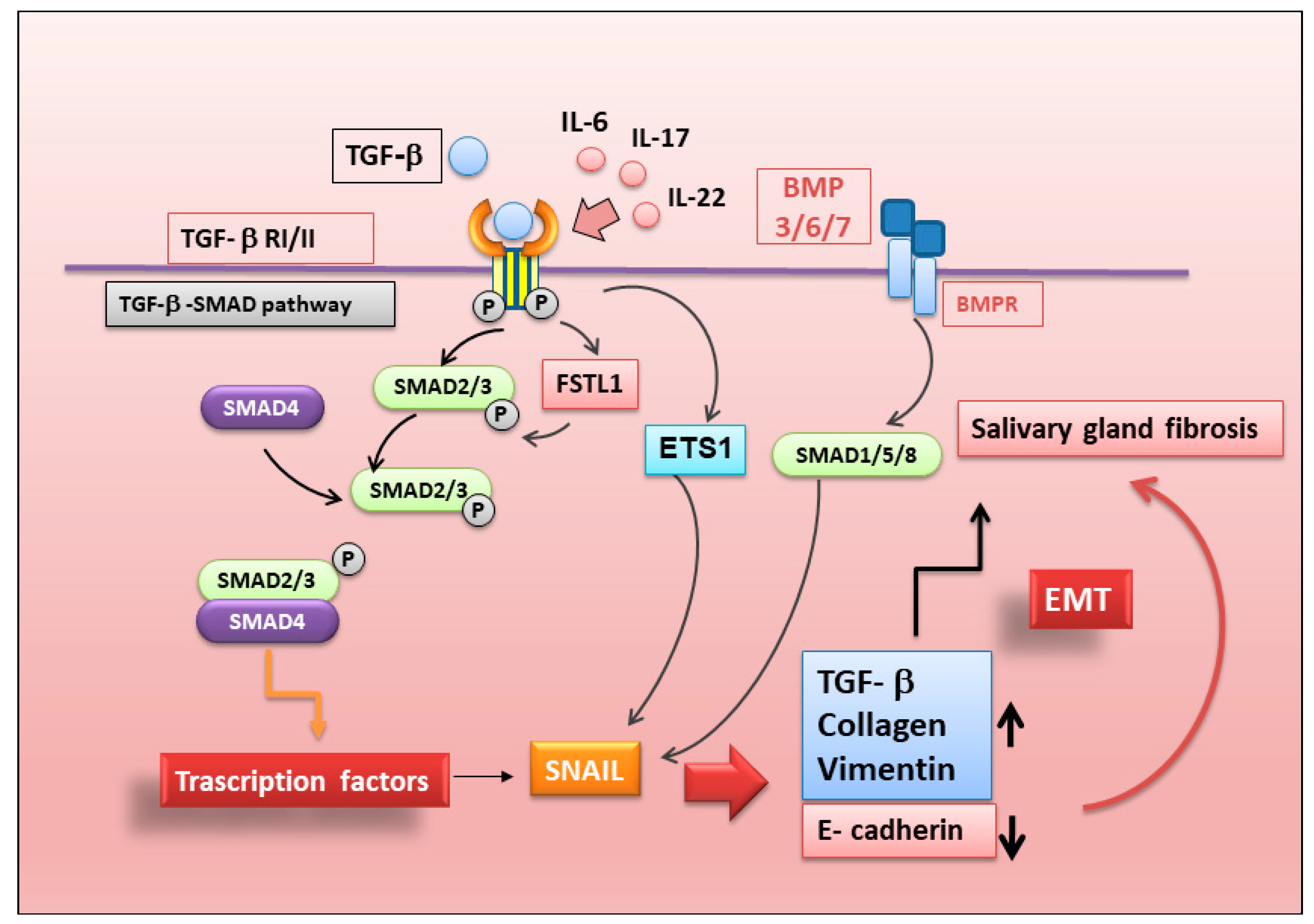
5. New Insights into the Regulation of Epithelial–Mesenchymal Transition in SS
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moutsopoulos, H.M. Sjögren’s syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 1994, 72, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Criswell, L.A. Primary Sjögren’s syndrome. N. Engl. J. Med. 2018, 378, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Skarlis, C.; Raftopoulou, S.; Mavragani, C.P. Sjogren’s Syndrome: Recent Updates. J. Clin. Med. 2022, 11, 399. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, X.; Zhang, C.; Su, M.; Gao, S.; Wang, J.; Lu, C.; Lin, Q.; Lin, J.; Matucci-Cerinic, M.; et al. Rheumatoid arthritis, systemic lupus erythematosus and primary Sjögren’s syndrome shared megakaryocyte expansion in peripheral blood. Ann. Rheum. Dis. 2022, 81, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.I.; McHugh, N.J. Apoptosis-new clues to the pathogenesis of Sjögren’s syndrome? Rheumatology 2000, 39, 119–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manganelli, P.; Fietta, P. Apoptosis and Sjögren syndrome. Semin. Arthritis Rheum. 2003, 33, 49–65. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Humphreys-Beher, M.G.; Peck, A.B.; Dang, H.; Talal, N. The role of apoptosis in the initiation of the autoimmune response in Sjögren’s syndrome. Clin. Exp. Immunol. 1999, 116, 383–387. [Google Scholar] [CrossRef]
- Božič, B.; Rozman, B. Apoptosis and Autoimmunity. EJIFCC 2006, 17, 69–74. [Google Scholar]
- Ogawa, Y.; Takeuchi, T.; Tsubota, K. Autoimmune Epithelitis and Chronic Inflammation in Sjögren’s Syndrome-Related Dry Eye Disease. Int. J. Mol. Sci. 2021, 22, 11820. [Google Scholar] [CrossRef]
- Shibata, Y.; Hishikawa, Y.; Izumi, S.; Fujita, S.; Yamaguchi, A.; Koji, T. Involvement of Fas/Fas ligand in the induction of apoptosis in chronic sialadenitis of minor salivary glands including Sjögren’s syndrome. Hum. Cell 2002, 15, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kawakami, A.; Izumi, M.; Nakashima, T.; Takagi, Y.; Ida, H.; Nakamura, T.; Nakamura, T.; Eguchi, K. Detection of the soluble form of Fas ligand (sFasL) and sFas in the saliva from patients with Sjögren’s syndrome. Clin. Exp. Rheumatol. 2005, 23, 915. [Google Scholar] [PubMed]
- Nakamura, H.; Horai, Y.; Shimizu, T.; Kawakami, A. Modulation of Apoptosis by Cytotoxic Mediators and Cell-Survival Molecules in Sjögren’s Syndrome. Int. J. Mol. Sci. 2018, 19, 2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haneji, N.; Nakamura, T.; Takio, K.; Yanagi, K.; Higashiyama, H.; Saito, I.; Noji, S.; Sugino, H.; Hayashi, Y. Identification of α-fodrin as a candidate autoantigen in primary Sjögren’s syndrome. Science 1997, 276, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Bulosan, M.; Pauley, K.M.; Yo, K.; Chan, E.K.; Katz, J.; Peck, A.B.; Cha, S. Inflammatory caspases are critical for enhanced cell death in the target tissue of Sjögren’s syndrome before disease onset. Immunol. Cell Biol. 2009, 87, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Casiano, C.A.; Martin, S.J.; Green, D.R.; Tan, E.M. Selective cleavage of nuclear autoantigens during CD95 (Fas/Apo-1)-mediated T cell apoptosis. J. Exp. Med. 1996, 184, 765–770. [Google Scholar] [CrossRef] [Green Version]
- Sandhya, P.; Kurien, B.T.; Danda, D.; Scofield, R.H. Update on Pathogenesis of Sjogren’s Syndrome. Curr. Rheumatol. Rev. 2017, 13, 5–22. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Font, J. Primary Sjögren’s syndrome: Current and emergent aetiopathogenic concepts. Rheumatology 2005, 44, 1354–1367. [Google Scholar] [CrossRef] [Green Version]
- Sisto, M.; Lisi, S.; Castellana, D.; Scagliusi, P.; D’Amore, M.; Caprio, S.; Scagliusi, A.; Acquafredda, A.; Panaro, M.A.; Mitolo, V. Autoantibodies from Sjögren’s syndrome induce activation of both the intrinsic and extrinsic apoptotic pathways in human salivary gland cell line A-253. J. Autoimmun. 2006, 27, 38–49. [Google Scholar] [CrossRef]
- Deets, K.A.; Vance, R.E. Inflammasomes and Adaptive Immune Responses. Nat. Immunol. 2021, 22, 412–422. [Google Scholar] [CrossRef]
- Hachim, M.Y.; Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Pyroptosis: The Missing Puzzle Among Innate and Adaptive Immunity Crosstalk. J. Leukoc. Biol. 2020, 108, 323–338. [Google Scholar] [CrossRef]
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging Insights into Molecular Mechanisms Underlying Pyroptosis and Functions of Inflammasomes in Diseases. J. Cell Physiol. 2020, 235, 3207–3221. [Google Scholar] [CrossRef] [PubMed]
- Vakrakou, A.G.; Boiu, S.; Ziakas, P.D.; Xingi, E.; Boleti, H.; Manoussakis, M.N. Systemic Activation of NLRP3 Inflammasome in Patients with Severe Primary Sjögren’s Syndrome Fueled by Inflammagenic DNA Accumulations. J. Autoimmun. 2018, 91, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Vakrakou, A.G.; Svolaki, I.P.; Evangelou, K.; Gorgoulis, V.G.; Manoussakis, M.N. Cell-Autonomous Epithelial Activation of AIM2 (Absent in Melanoma-2) Inflammasome by Cytoplasmic DNA Accumulations in Primary Sjögren’s Syndrome. J. Autoimmun. 2020, 108, 102381. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.M.; Lee, J.; Jang, S.G.; Lee, J.; Cho, M.L.; Kwok, S.K.; Park, S.H. Type I Interferon Increases Inflammasomes Associated Pyroptosis in the Salivary Glands of Patients with Primary Sjögren’s Syndrome. Immune. Netw. 2020, 20, e39. [Google Scholar] [CrossRef] [PubMed]
- Church, J.A. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Pediatrics 2014, 34, S184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Chu, A. Sjögren’s Syndrome and Viral Infections. Rheumatol. Ther. 2021, 8, 1051–1059. [Google Scholar] [CrossRef]
- Nakamura, H.; Shimizu, T.; Kawakami, A. Role of Viral Infections in the Pathogenesis of Sjögren’s Syndrome: Different Characteristics of Epstein-Barr Virus and HTLV-1. J. Clin. Med. 2020, 9, 1459. [Google Scholar] [CrossRef]
- Ishimaru, N.; Arakaki, R.; Omotehara, F.; Yamada, K.; Mishima, K.; Saito, I.; Hayashi, Y. Novel role for RbAp48 in tissue-specific, estrogen deficiency-dependent apoptosis in the exocrine glands. Mol. Cell. Biol. 2006, 26, 2924–2935. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, N.; Arakaki, R.; Yoshida, S.; Yamada, A.; Noji, S.; Hayashi, Y. Expression of the retinoblastoma protein RbAp48 in exocrine glands leads to Sjögren’s syndrome-like autoimmune exocrinopathy. J. Exp. Med. 2008, 205, 2915–2927. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, K.; Sato, M.; Tsunematsu, T.; Ishimaru, N. Virus Infections Play Crucial Roles in the Pathogenesis of Sjögren’s Syndrome. Viruses 2022, 14, 1474. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Warner, B.M.; Odani, T.; Ji, Y.; Mo, Y.Q.; Nakamura, H.; Jang, S.I.; Yin, H.; Michael, D.G.; Hirata, N.; et al. LAMP3 induces apoptosis and autoantigenrelease in Sjögren’s syndrome patients. Sci. Rep. 2020, 10, 15169. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Tanaka, T.; Pranzatelli, T.; Ji, Y.; Yin, H.; Perez, P.; Afione, S.A.; Jang, S.I.; Goldsmith, C.; Zheng, C.Y.; et al. Lysosome-associated membrane protein 3 misexpression in salivary glands induces a Sjögren’s syndrome-like phenotype in mice. Ann. Rheum. Dis. 2021, 80, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Crivellato, E. Sprouting angiogenesis, a reappraisal. Dev. Biol. 2012, 15, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Majno, G. Chronic inflammation: Links with angiogenesis and wound healing. Am. J. Pathol. 1998, 153, 1035–1039. [Google Scholar] [CrossRef]
- Bagli, E.; Xagorari, A.; Papetropoulos, A.; Murphy, C.; Fotsis, T. Angiogenesis in inflammation. Autoimmun. Rev. 2004, 3, S26. [Google Scholar]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; D’Amore, M.; Frassanito, M.A.; Ribatti, D. Sjögren’s syndrome pathological neovascularization is regulated by VEGF-A-stimulated TACE-dependent crosstalk between VEGFR2 and NF-κB. Genes Immun. 2012, 13, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Abu-Helu, R.F.; Dimitriou, I.D.; Kapsogeorgou, E.K.; Moutsopoulos, H.M.; Manoussakis, M.N. Induction of salivary gland epithelial cell injury in Sjogren’s syndrome: In vitro assessment of T cell-derived cytokines and Fas protein expression. J. Autoimmun. 2001, 17, 141–153. [Google Scholar] [CrossRef]
- Gerli, R.; Vaudo, G.; Bocci, E.B. Functional impairment of the arterial wall in primary Sjögren’s syndrome: Combined action of immunologic and inflammatory factors. Arthritis Care Res. 2010, 62, 712–718. [Google Scholar] [CrossRef]
- Thakor, A.S.; Brown, C.N.; Edwards, A. Effects of prolonged reduction in blood flow on submandibular secretory function in anesthetized sheep. J. Appl. Physiol. 2003, 95, 751–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; D’Amore, M.; Ribatti, D. Neuropilin-1 is upregulated in Sjögren’s syndrome and contributes to pathological neovascularization. Histochem. Cell Biol. 2012, 137, 669–677. [Google Scholar] [CrossRef] [PubMed]
- McCall, A.D.; Baker, O.J. Characterization of Angiogenesis and Lymphangiogenesis in Human Minor Salivary Glands with Sjögren’s Syndrome. J. Histochem. Cytochem. 2015, 63, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alunno, A.; Ibba-Manneschi, L.; Bistoni, O.; Cipriani, S.; Topini, F.; Gerli, R.; Manetti, M. Angiogenic T cells in primary Sjögren’s syndrome: A double-edged sword? Clin. Exp. Rheumatol. 2019, 118, 36–41. [Google Scholar]
- Bartoloni, E.; Alunno, A.; Bistoni, O.; Caterbi, S.; Luccioli, F.; Santoboni, G.; Mirabelli, G.; Cannarile, F.; Gerli, R. Characterization of circulating endothelial microparticles and endothelial progenitor cells in primary Sjögren’s syndrome: New markers of chronic endothelial damage? Rheumatology 2015, 54, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, J.; Yang, H.M.; Yoon, C.H.; Lee, C.S.; Park, K.W.; Kim, J.H.; Kim, T.Y.; Kim, J.Y.; Kang, H.J.; Chae, I.H.; et al. Identification of a novel role of T cells in postnatal vasculogenesis: Characterization of endothelial progenitor cell colonies. Circulation 2007, 116, 1671–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisto, M.; Lorusso, L.; Tamma, R.; Ingravallo, G.; Ribatti, D.; Lisi, S. Interleukin-17 and -22 synergy linking inflammation and EMT-dependent fibrosis in Sjögren’s syndrome. Clin. Exp. Immunol. 2019, 198, 261–272. [Google Scholar] [CrossRef]
- Cannarile, F.; Valentini, V.; Mirabelli, G.; Alunno, A.; Terenzi, R.; Luccioli, F.; Gerli, R.; Bartoloni, E. Cardiovascular disease in systemic sclerosis. Ann. Transl. Med. 2015, 3, 8. [Google Scholar] [PubMed]
- Vaudo, G.; Bocci, E.B.; Shoenfeld, Y.; Schillaci, G.; Wu, R.; Del Papa, N.; Vitali, C.; Delle Monache, F.; Marchesi, S.; Mannarino, E.; et al. Precocious intima-media thickening in patients with primary Sjögren’s syndrome. Arthritis Rheum. 2015, 52, 3890–3897. [Google Scholar] [CrossRef]
- Thomson, L. 3-Nitrotyrosine modified proteins in atherosclerosis. Dis. Markers 2015, 2015, 708282. [Google Scholar] [CrossRef]
- Soyfoo, M.S.; Chivasso, C.; Perret, J.; Delporte, C. Involvement of Aquaporins in the Pathogenesis, Diagnosis and Treatment of Sjögren’s Syndrome. Int. J. Mol. Sci. 2018, 19, 3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agre, P. Aquaporin water channels (Nobel lecture). Angew Chem. Int. Ed. Engl. 2004, 43, 4278–4290. [Google Scholar] [CrossRef] [PubMed]
- Delporte, C. Aquaporins and Gland Secretion. Adv. Exp. Med. Biol. 2017, 969, 63–79. [Google Scholar] [PubMed]
- Delporte, C.; Bryla, A.; Perret, J. Aquaporins in Salivary Glands: From Basic Research to Clinical Applications. Int. J. Mol. Sci. 2016, 17, 166. [Google Scholar] [CrossRef] [Green Version]
- Ichiyama, T.; Nakatani, E.; Tatsumi, K.; Hideshima, K.; Urano, T.; Nariai, Y.; Sekine, J. Expression of aquaporin 3 and 5 as a potential marker for distinguishing dry mouth from Sjogren’s syndrome. J. Oral Sci. 2018, 60, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Delporte, C.; Soyfoo, M. Aquaporins: Unexpected actors in autoimmune diseases. Autoimmun. Rev. 2022, 21, 103131. [Google Scholar] [CrossRef]
- Beroukas, D.; Hiscock, J.; Jonsson, R.; Waterman, S.A.; Gordon, T.P. Subcellular distribution of aquaporin 5 in salivary glands in primary Sjogren’s syndrome. Lancet 2001, 358, 1875–1876. [Google Scholar] [CrossRef]
- Delporte, C.; Steinfeld, S. Distribution and roles of aquaporins in salivary glands. Biochim. Biophys. Acta 2006, 1758, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Nico, B.; Ribatti, D.; Ruggieri, S.; Lofrumento, D.D.; Lisi, S. Abnormal distribution of AQP4 in minor salivary glands of primary Sjogren’s syndrome patients. Autoimmunity 2017, 50, 202–210. [Google Scholar] [CrossRef]
- Steinfeld, S.; Cogan, E.; King, L.S.; Agre, P.; Kiss, R.; Delporte, C. Abnormal distribution of aquaporin-5 water channel protein in salivary glands from Sjogren’s syndrome patients. Lab. Investig. 2001, 81, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Meli, R.; Pirozzi, C.; Pelagalli, A. New Perspectives on the Potential Role of Aquaporins (AQPs) in the Physiology of Inflammation. Front. Physiol. 2018, 9, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, I.V.; Soveral, G. Aquaporins in Immune Cells and Inflammation: New Targets for Drug Development. Int. J. Mol. Sci. 2021, 22, 1845. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, Q. Anti-IL-7 receptor-alpha treatment ameliorates newly established Sjogren’s-like exocrinopathy in non-obese diabetic mice. Biochim. Biophys. Acta 2018, 1864, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kawai, T.; Yu, Q. Pathogenic role of endogenous TNF-alpha in the development of Sjogren’s-like sialadenitis and secretory dysfunction in non-obese diabetic mice. Lab. Investig. 2017, 97, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Koh, J.H.; Kim, N.; Kwok, S.K.; Park, S.H.; Song, Y.W.; Park, K.; Choi, Y. Detection of autoantibodies against aquaporin-5 in the sera of patients with primary Sjogren’s syndrome. Immunol. Res. 2016, 64, 848–856. [Google Scholar] [CrossRef] [Green Version]
- Tzartos, J.S.; Stergiou, C.; Daoussis, D.; Zisimopoulou, P.; Andonopoulos, A.P.; Zolota, V.; Tzartos, S.J. Antibodies to aquaporins are frequent in patients with primary Sjogren’s syndrome. Rheumatology 2017, 56, 2114–2122. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-Mesenchymal Transition and Its Implications for Fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leehan, K.M.; Pezant, N.P.; Rasmussen, A.; Grundahl, K.; Moore, J.S.; Radfar, L.; Lewis, D.M.; Stone, D.U.; Lessard, C.J.; Rhodus, N.L.; et al. Minor salivary gland fibrosis in Sjögren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin. Exp. Rheumatol. 2018, 112, 80–88. [Google Scholar]
- Lendrem, D.; Mitchell, S.; McMeekin, P.; Bowman, S.; Price, E.; Pease, C.T.; Emery, P.; Andrews, J.; Lanyon, P.; Hunter, J.; et al. Health-related utility values of patients with primary Sjogren’s syndrome and its predictors. Ann. Rheum. Dis. 2014, 73, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bottinger, E.P. TGF-β and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef]
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Ribatti, D.; Lisi, S. TGFβ1-Smad canonical and -Erk non-canonical pathways participate in interleukin-17-induced epithelial–mesenchymal transition in Sjögren’s syndrome. Lab. Investig. 2020, 100, 824–836. [Google Scholar] [CrossRef]
- Koski, H.; Konttinen, Y.T.; Gu, X.H.; Hietanen, J.; Malmström, M. Transforming growth factor beta 2 in labial salivary glands in Sjögren’s syndrome. Ann. Rheum. Dis. 1995, 54, 744–747. [Google Scholar] [CrossRef] [Green Version]
- Sisto, M.; Tamma, R.; Ribatti, D.; Lisi, S. IL-6 Contributes to the TGF-β1-Mediated Epithelial to Mesenchymal Transition in Human Salivary Gland Epithelial Cells. Arch. Immunol. Ther. Exp. 2020, 68, 27. [Google Scholar] [CrossRef]
- Sisto, M.; Ribatti, D.; Lisi, S. Cadherin Signaling in Cancer and Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 13358. [Google Scholar] [CrossRef]
- Yin, H.; Kalra, L.; Lai, Z.; Guimaro, M.C.; Aber, L.; Warner, B.M.; Michael, D.; Zhang, N.; Cabrera-Perez, J.; Karim, A.; et al. Inhibition of Bone Morphogenetic Protein 6 Receptors Ameliorates Sjogren’s Syndrome in Mice. Sci. Rep. 2020, 10, 2967. [Google Scholar] [CrossRef] [Green Version]
- Noll, B.; Mougeot, F.B.; Brennan, M.T.; Mougeot, J.C. Regulation of MMP9 transcription by ETS1 in immortalized salivary gland epithelial cells of patients with salivary hypofunction and primary Sjögren’s syndrome. Sci. Rep. 2022, 12, 14552. [Google Scholar] [CrossRef]
- Liu, T.; Liu, Y.; Miller, M.; Cao, L.; Zhao, J.; Wu, J.; Wang, J.; Liu, L.; Li, S.; Zou, M.; et al. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L27–L40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Qi, C.; Zhang, S.; Fang, Y.; Ning, W. TGF-β1 induces Fstl1 via the Smad3-c-Jun pathway in lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L240–L251. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Geng, Y.; Li, L.; Li, X.; Yan, X.; Fang, Y.; Li, X.; Dong, S.; Liu, X.; Li, X.; et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J. Exp. Med. 2015, 212, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Ribatti, D.; Ingravallo, G.; Lisi, S. The Expression of Follistatin-like 1 Protein Is Associated with the Activation of the EMT Program in Sjögren’s Syndrome. J. Clin. Med. 2022, 11, 5368. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nomenclatures of Therapies | Definition | Examples |
|---|---|---|
| Thopical therapies | Interventions directly applied to the mucosal surface involved | Saliva substitutes, ocular tears, ocular gels/ointment |
| Systemic therapies | Drugs administered orally or intravenously for systemic disease | Antimalarials, glucocorticoids, immunosuppressive agents, intravenous immunoglobulins, biologics drugs |
| Systemic therapies for severe refractory diseases | Drugs administered intravenously | B-cells targeted therapies |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sisto, M.; Ribatti, D.; Lisi, S. Molecular Mechanisms Linking Inflammation to Autoimmunity in Sjögren’s Syndrome: Identification of New Targets. Int. J. Mol. Sci. 2022, 23, 13229. https://doi.org/10.3390/ijms232113229
Sisto M, Ribatti D, Lisi S. Molecular Mechanisms Linking Inflammation to Autoimmunity in Sjögren’s Syndrome: Identification of New Targets. International Journal of Molecular Sciences. 2022; 23(21):13229. https://doi.org/10.3390/ijms232113229
Chicago/Turabian StyleSisto, Margherita, Domenico Ribatti, and Sabrina Lisi. 2022. "Molecular Mechanisms Linking Inflammation to Autoimmunity in Sjögren’s Syndrome: Identification of New Targets" International Journal of Molecular Sciences 23, no. 21: 13229. https://doi.org/10.3390/ijms232113229