
What Are the Reasons for Continuing Failures in Cancer Therapy? Are Misleading/Inappropriate Preclinical Assays to Be Blamed? Might Some Modern Therapies Cause More Harm than Benefit?
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: Why Is the ‘War on Cancer’ Not Won Yet?
2. Well Established and Yet Widely Overlooked Therapy-Induced Cancer Cell Responses That Contribute to Disease Recurrence
2.1. Cell Recovery from Death through Anastasis
2.2. Pro-Survival Functions of Caspase-3
2.3. Impact of Therapy-Induced Cancer Cell Dormancy through Polyploidy, Multinucleation and/or Premature Senescence on Disease Recurrence
- PGCCs are present in solid tumors/tumor-derived cell lines with differing p53 status, and their frequency typically increases under hypoxia or following treatment with anticancer agents [50]. Non-genotoxic agents are also known to promote the creation of PGCCs. These include nutlin-3a [64], a small molecule activator of wild-type p53, and staurosporine [65], which is commonly used as a standard apoptosis-inducing agent.
- PGCCs can be created through endoreduplication (replication of chromosomes without subsequent cell division) and cell fusion [4]. Fusions between cancer cells and cancer cells, cancer cells and leukocytes, cancer cells and stem cells, and cancer cells and stromal cells are all known to promote tumor progression and therapy resistance [4].
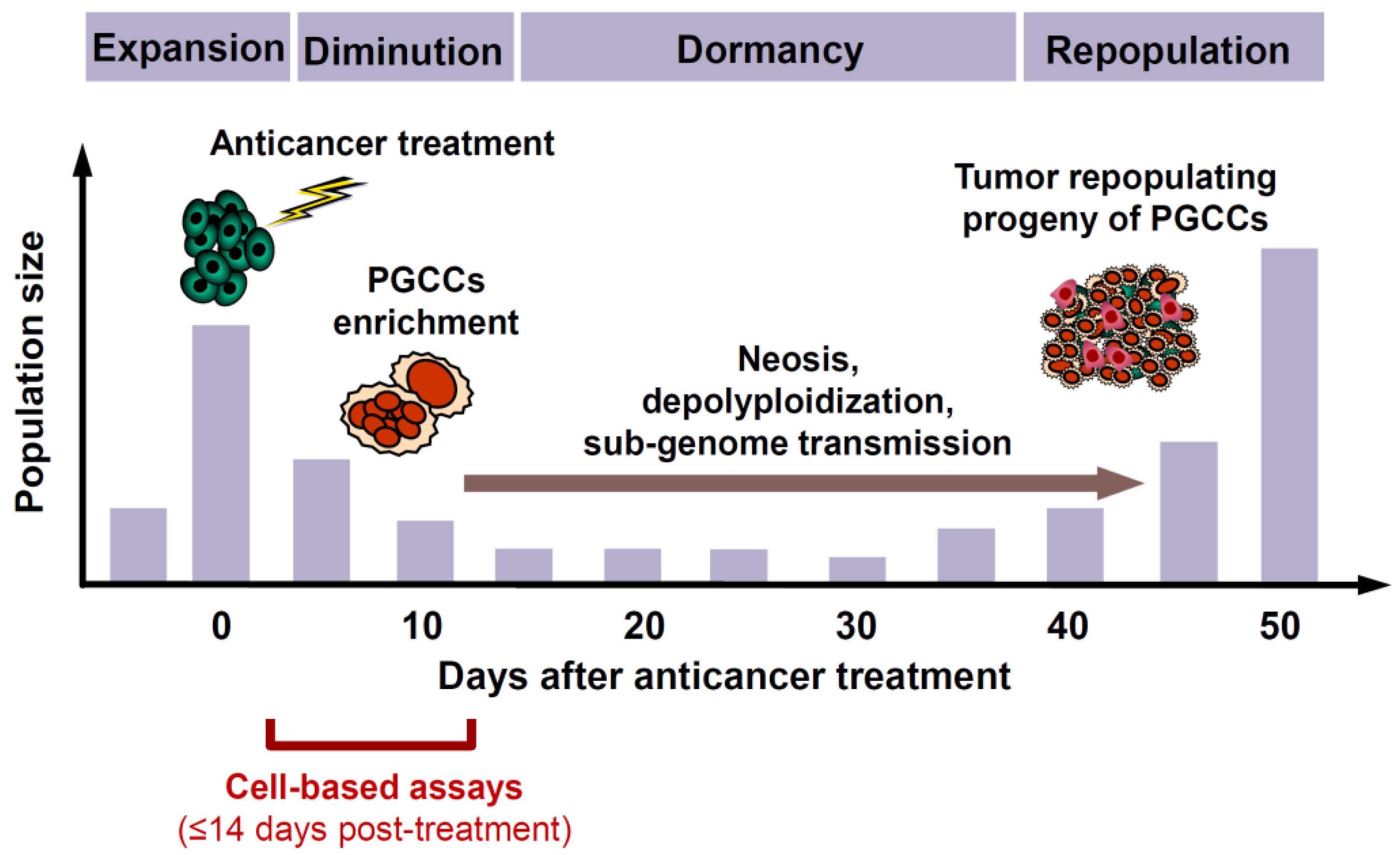
- Although PGCCs created following treatment with anticancer agents initially (within the time span of conventional preclinical assays) cease to proliferate, they remain viable and metabolically active [50].
- Subsequently, PGCCs can not only undergo depolyploidization and resume mitosis, but they are also capable of giving rise to therapy-resistant and tumor repopulating cells through processes such as neosis (nuclear budding or bursting) as well as horizontal transfer of their nuclear material that contains stem-cell markers to neighboring cells [50].
- Although PGCCs are often fully manifested within ~3 days after treatment with anticancer agents and their depolyploidization and nuclear budding processes can commence at any time thereafter, it can take several weeks or months until a stable, rapidly proliferating population of daughter cells emerges that repopulates the tumor [50]. Thus, as illustrated in Figure 1, the time required between therapeutic exposure and emergence of tumor repopulating progeny of cancer cells triggered to undergoing dormancy through polyploidy and/or multinucleation is much longer than the time span of multiwell plate cell “viability” (e.g., MTT, CellTiterGlo, etc.), colony formation, and other widely used preclinical anticancer assays for cell “killing”.
3. Snapshot of the History of Cancer Research
3.1. Strengths and Limitations of the Reductionist Approach to Cancer Biology
3.2. Publishing “Mansions of Straw” versus “House of Brick” Research Papers
- Danger of cramming large amounts of data into a single manuscript, which often includes extensive data sets presented in the supplementary material, without taking the time to scrutinize what the results of each assay actually indicate. Kaelin uses the analogy of “building with straw” for such publications and states that the “real advances are built with bricks, not straw” [104].
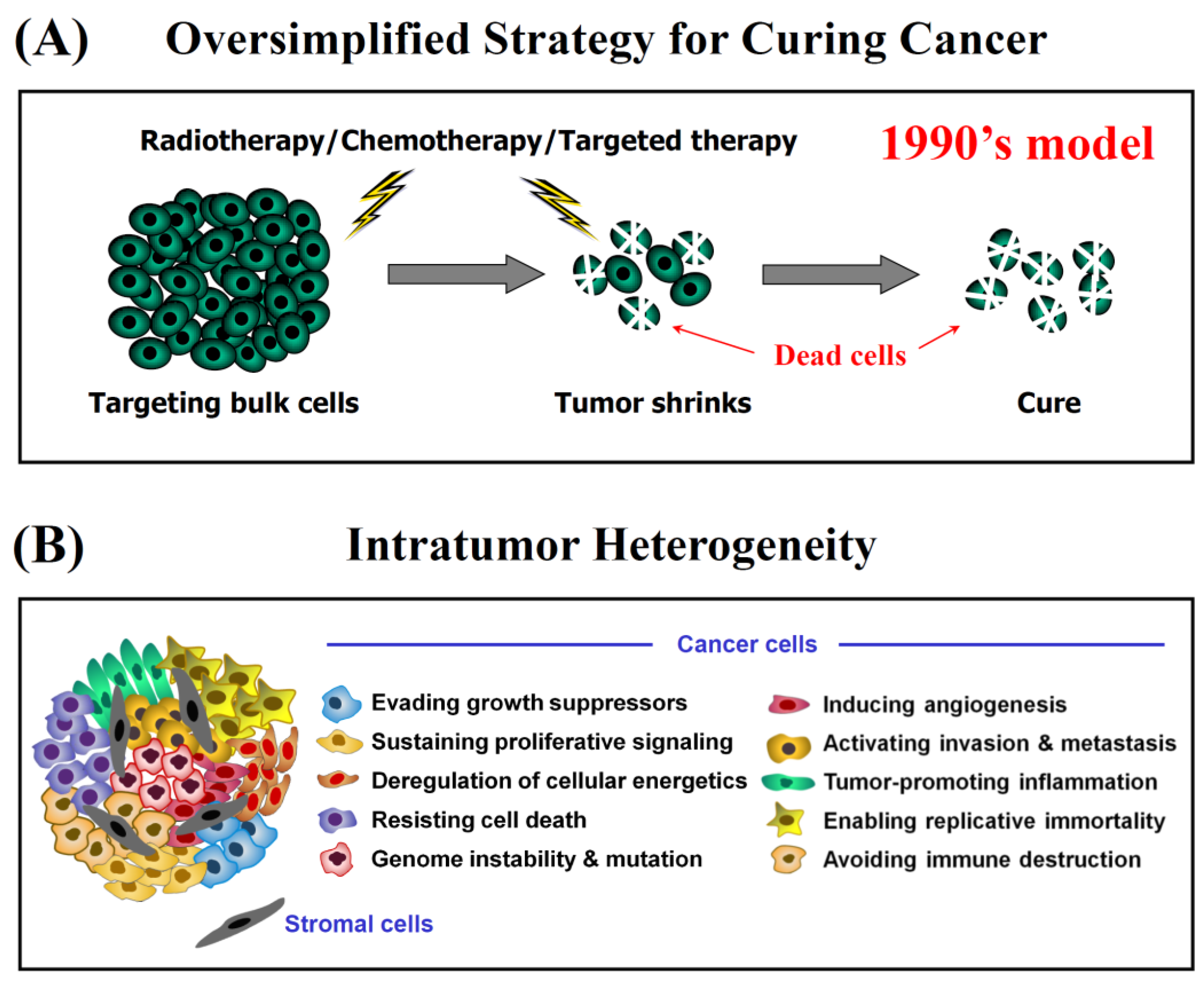
- Danger of jumping to conclusions from ubiquitous use of “down” assays (e.g., decreased proliferation, decreased tumor growth, etc.) to clinical relevance. For example, as we see in numerous recent articles, if a test drug is shown to inhibit proliferation (e.g., in potentially highly misleading multiwell plate assays; see, e.g., [4,18]), down-regulate global levels of the protein under study in immunoblot analysis, and inhibit tumor growth in live (often immune deficient) animals, it is typically concluded that the test drug must have clinical relevance as a novel anticancer agent when administered with or without conventional therapies.
- Potential bias/misconception that can arise from the Kaplan–Meier survival curves and tumor growth delay assays in live animals when used to analyze prognostic/predictive molecular biomarkers of tumor response. Kaelin indicated that he sometimes calls this “the marriage of the gratuitous and the contrived” [105].
3.3. Off-Target Effects of Anticancer Drugs Developed for Targeted Therapies
3.4. Cancer Immunoediting
4. Dishonesty in Reporting Data Due to Pressure to Publish, and Not Reporting the Scientific Bases of Failed Clinical Outcomes for the Benefit of Drug Manufacturers
5. Do Modern Cancer Therapies Prolong Survival of Patients with Solid Tumors?
6. Impact of Surgery on Tumor Progression
7. Where Do We Go from Here in the War on Cancer?
- Regarding cancer as a network phenomenon (e.g., [145,146,147,148]). To this end, as recently pointed out by Erenpreisa and Giuliani [147], “The reductionist, one gene/one protein method that has served us well until now, and that still dominates in biomedicine, requires complementation with a more systemic/holistic approach, to address the huge problem of cross-talk between more than 20,000 protein-coding genes, about 100,000 protein types, and the multiple layers of biological organization.”
- Abandoning the use of immune deficient/compromised animal models in anticancer-related studies, or at least being aware of their limitations (see below).
8. Conclusions
Funding
Conflicts of Interest
Appendix A

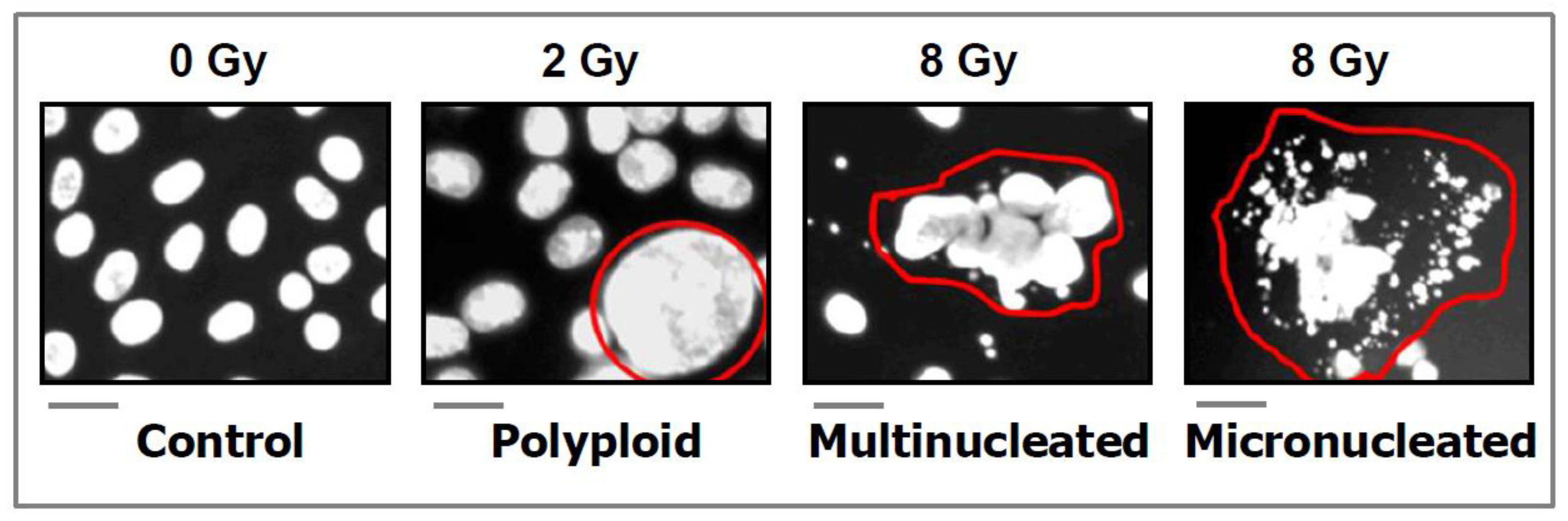
- DAPI (4′:6-diamidino-2-phenylindole) images showing the development of polyploid, multinucleated, and micronucleated giant cells in the SUM159 breast carcinoma cell line after exposure to ionizing radiation and incubation for 3 days. Scale bars, 20 µm. Reproduced from Mirzayans and Murray [26].
Appendix B

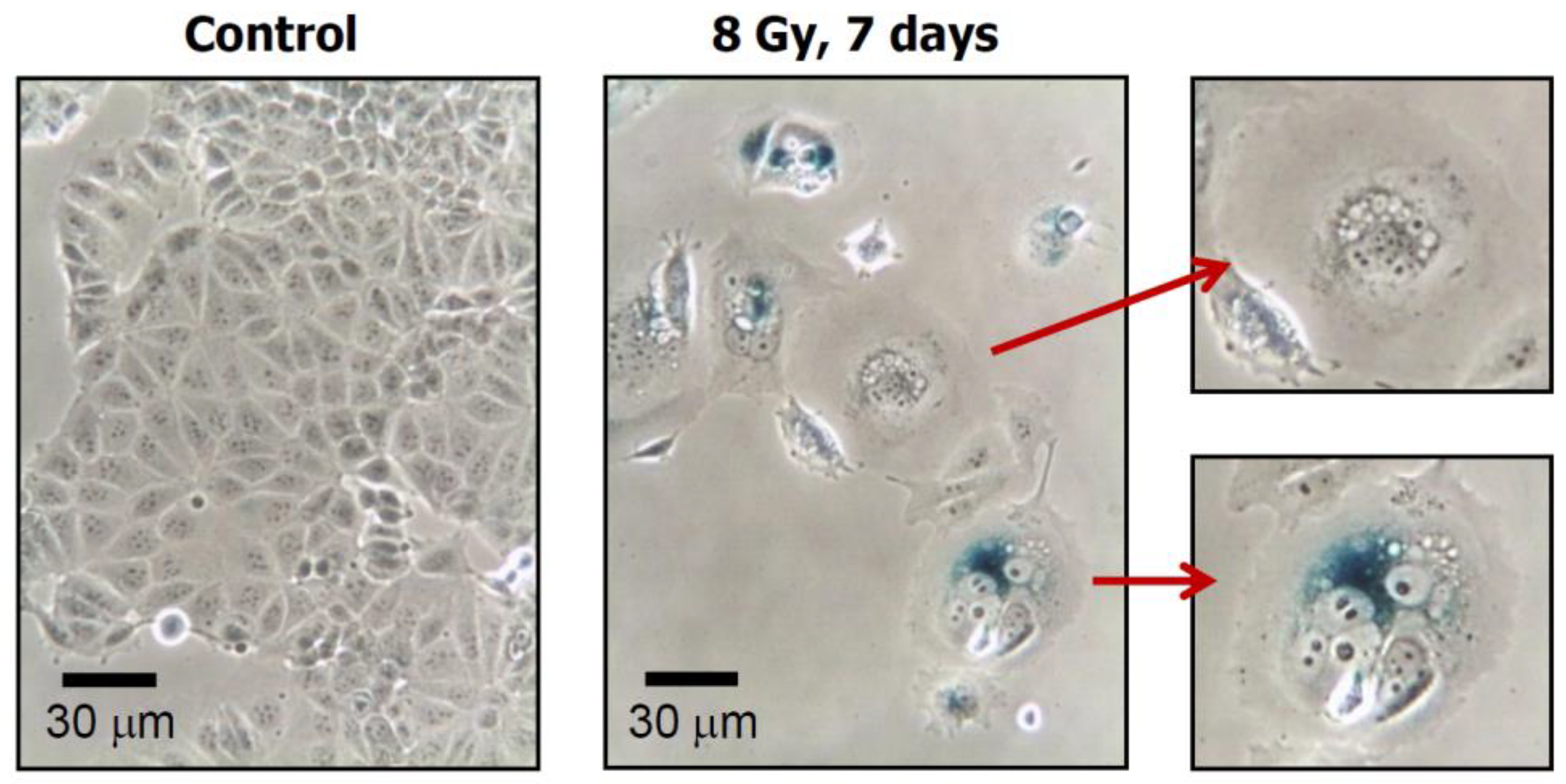
- Phase-contrast photomicrographs showing premature senescence in MDD2, a mutant p53-expressing derivative of the MCF7 breast carcinoma cell line. Cultures were exposed to ionizing radiation (8 Gy) or sham-irradiated (control), incubated for seven days, and evaluated for flattened and enlarged cellular morphology and positive (blue) staining in the senescence-associated β-galactosidase assay. Reproduced from Mirzayans et al. [50].
References
- Editorial. The ‘war on cancer’ isn’t yet won. Nature 2022, 601, 297. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and therapy resistance: Contributions of dormancy, apoptosis reversal (anastasis) and cell fusion to disease recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [Green Version]
- Geske, F.J.; Lieberman, R.; Strange, R.; Gerschenson, L.E. Early stages of p53-induced apoptosis are reversible. Cell Death Differ. 2001, 8, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Geske, F.J.; Nelson, A.C.; Lieberman, R.; Strange, R.; Sun, T.; Gerschenson, L.E. DNA repair is activated in early stages of p53-induced apoptosis. Cell Death Differ. 2000, 7, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return journey from cell death. Cancers 2021, 13, 3671. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Fung, M.C.; Hardwick, J.M. In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci. Rep. 2015, 5, 9015. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.L.; Yuen, K.L.; Tang, H.M.; Fung, M.C. Reversibility of apoptosis in cancer cells. Br. J. Cancer 2009, 100, 118–122. [Google Scholar] [CrossRef]
- Tang, H.M.; Talbot, C.C., Jr.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000Research 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seervi, M.; Sumi, S.; Chandrasekharan, A.; Sharma, A.K.; SanthoshKumar, T.R. Molecular profiling of anastatic cancer cells: Potential role of the nuclear export pathway. Cell Oncol. 2019, 42, 645–661. [Google Scholar] [CrossRef]
- Wang, S.S.; Xie, X.; Wong, C.S.; Choi, Y.; Fung, M.C. HepG2 cells recovered from apoptosis show altered drug responses and invasiveness. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 293–300. [Google Scholar] [CrossRef]
- Xu, Y.; So, C.; Lam, H.M.; Fung, M.C.; Tsang, S.Y. Flow cytometric detection of newly-formed breast cancer stem cell-like cells after apoptosis reversal. J. Vis. Exp. 2019, 143, e58642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; So, C.; Lam, H.-M.; Fung, M.-C.; Tsang, S.-Y. Apoptosis reversal promotes cancer stem cell-like cell formation. Neoplasia 2018, 20, 295–303. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Hardwick, J.M.; Fung, M.C. Strategies for tracking anastasis, a cell survival phenomenon that reverses apoptosis. J. Vis. Exp. 2015, 96, e51964. [Google Scholar] [CrossRef] [Green Version]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [Google Scholar] [CrossRef] [Green Version]
- Nicoletto, R.E.; Ofner, C.M., III. Cytotoxic mechanisms of doxorubicin at clinically relevant concentrations in breast cancer cells. Cancer Chemother. Pharmacol. 2022, 89, 285–311. [Google Scholar] [CrossRef]
- Gupta, S.; Kass, G.E.N.; Szegezdi, E.; Joseph, B. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033. [Google Scholar] [CrossRef]
- Ichim, G.; Lopez, L.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, S.J.; Mason, S.M.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.M.; Tang, H.L. Anastasis: Recovery from the brink of cell death. R. Soc. Open Sci. 2018, 5, 180442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.-N.; Crawford, J.C.; Heckmann, B.L.; Green, D.R. To the edge of cell death and back. FEBS J. 2019, 286, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional ways to live and die: Cell death and survival in development, homeostasis, and disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, I.I.; Savitskaya, M.A.; Onishchenko1, G.E. The problem of apoptotic processes reversibility. Biochemistry 2020, 85, 1145–1158. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Do TUNEL and other apoptosis assays detect cell death in preclinical studies? Int. J. Mol. Sci. 2020, 21, 9090. [Google Scholar] [CrossRef]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.-T.; Waseem, A. Vimentin is at the heart of epithelial mesenchymal transition (EMT) mediated metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef]
- Mohammed, R.N.; Khosravi, M.; Rahman, H.S.; Adili, A.; Kamali, N.; Soloshenkov, P.P.; Thangavelu, L.; Saeedi, H.; Shomali, N.; Tamjidifar, R.; et al. Anastasis: Cell recovery mechanisms and potential role in cancer. Cell Commun. Signal. 2022, 20, 81. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.Y. Apoptotic cells activate the ‘‘phoenix rising’’ pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.-L.; Huang, Q.; Liu, X.; Zimmerman, M.; Li, F.; Li, C.-Y. Molecular mechanisms involved in tumor repopulation after radiotherapy. Transl. Cancer Res. 2013, 2, 442–448. [Google Scholar]
- Cheng, J.; Tian, L.; Ma, J.; Gong, Y.; Zhang, Z.; Chen, Z.; Xu, B.; Xiong, H.; Li, C.; Huang, Q. Dying tumor cells stimulate proliferation of living tumor cells via caspase-dependent protein kinase Cδ activation in pancreatic ductal adenocarcinoma. Mol. Oncol. 2015, 9, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.-Y. Caspase-3 promotes genetic instability and carcinogenesis. Mol. Cell 2015, 58, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, C.E.; Bergmann, A. Killers creating new life: Caspases drive apoptosis induced proliferation in tissue repair and disease. Cell Death Differ. 2017, 24, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartwright, I.M.; Liu, X.; Zhou, M.; Li, F.; Li, C.-Y. Essential roles of Caspase-3 in facilitating Myc-induced genetic instability and carcinogenesis. eLife 2017, 6, e26371. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, R.; Hur, J.; Kim, Y.J.; Kim, J.; Chwae, Y.J. Apoptotic cell-derived exosomes: Messages from dying cells. Exp. Mol. Med. 2020, 52, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Donato, A.L.; Huang, Q.; Liu, X.; Li, F.; Zimmerman, M.A.; Li, C.Y. Caspase 3 promotes surviving melanoma tumor cell growth after cytotoxic therapy. J. Investig. Dermatol. 2014, 134, 1686–1692. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, L.; Meyer, M.; Fay, J.; Curry, S.; Bacon, O.; Duessmann, H.; John, K.; Boland, K.C.; McNamara, D.A.; Kay, E.W.; et al. Low levels of caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: Caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016, 7, e2087. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Yu, Y.; He, S.; Cheng, J.; Gong, Y.; Zhang, Z.; Yang, X.; Xu, B.; Liu, X.; Li, C.Y.; et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 2017, 385, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.Y. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int. J. Cancer 2018, 143, 921–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, F.; Huang, Q.; Zhang, Z.; Zhou, L.; Deng, Y.; Zhou, M.; Fleenor, D.E.; Wang, H.; Kastan, M.B.; et al. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res. 2017, 27, 764–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Kaakati, R.; Lee, A.K.; Liu, X.; Li, F.; Li, C.-Y. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018, 37, 227–236. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, J.M.; Kim, J.; Hur, J.; Park, S.; Kim, K.; Shin, H.-J.; Chwae, Y.-J. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc. Natl. Acad. Sci. USA 2018, 115, E11721–E11730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsi, F.; Capradossi, F.; Pelliccia, A.; Briganti, S.; Bruni, E.; Traversa, E.; Torino, F.; Reichle, A.; Ghibelli, L. Apoptosis as driver of therapy-induced cancer repopulation and acquired cell-resistance (CRAC): A simple in vitro model of Phoenix Rising in prostate cancer. Int. J. Mol. Sci. 2022, 23, 1152. [Google Scholar] [CrossRef]
- Ichim, G.; Tait, S.W. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Berthenet, K.; Castillo Ferrer, C.; Fanfone, D.; Popgeorgiev, N.; Neves, D.; Bertolino, P.; Gibert, B.; Hernandez-Vargas, H.; Ichim, G. Failed apoptosis enhances melanoma cancer cell aggressiveness. Cell Rep. 2020, 31, 107731. [Google Scholar] [CrossRef]
- Castillo Ferrer, C.; Berthenet, K.; Ichim, G. Apoptosis—Fueling the oncogenic fire. FEBS J. 2021, 288, 4445–4463. [Google Scholar] [CrossRef]
- Herbein, G.; Nehme, Z. Polyploid giant cancer cells, a hallmark of oncoviruses and a new therapeutic challenge. Front. Oncol. 2020, 10, 567116. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of polyploid/multinucleated giant cancer cells in metastasis and disease relapse following anticancer treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, D.; Yang, Z.; Zhang, X. Tumor budding, micropapillary pattern, and polyploidy giant cancer cells in colorectal cancer: Current status and future prospects. Stem Cells Int. 2016, 2016, 4810734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20. [Google Scholar]
- Shu, Z.; Row, S.; Deng, W.M. Endoreplication: The good, the bad, and the ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef]
- Liu, J. The dualistic origin of human tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid Giant Cancer Cells (PGCCs): The evil roots of cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef]
- Ye, J.C.; Horne, S.; Zhang, J.Z.; Jackson, L.; Heng, H.H. Therapy induced genome chaos: A novel mechanism of rapid cancer drug resistance. Front. Cell Dev. Biol. 2021, 9, 676344. [Google Scholar] [CrossRef]
- Murray, D.; Mirzayans, R. Cellular responses to platinum-based anticancer drugs and UVC: Role of p53 and implications for cancer therapy. Int. J. Mol. Sci. 2020, 21, 5766. [Google Scholar] [CrossRef]
- Sikora, E.; Czarnecka-Herok, J.; Bojko, A.; Sunderland, P. Therapy-induced polyploidization and senescence: Coincidence or interconnection? Semin. Cancer Biol. 2022, 81, 83–95. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, Y.; Deng, Z.; Zhao, R.; Huang, Q. Stress-induced polyploid giant cancer cells: Unique way of formation and non-negligible characteristics. Front. Oncol. 2021, 11, 724781. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Hammarlund, E.U.; Brown, J.S.; Amend, S.R.; Axelrod, R.M. Cancer recurrence and lethality are enabled by enhanced survival and reversible cell cycle arrest of polyaneuploid cells. Proc. Natl. Acad. Sci USA 2021, 118, e2020838118. [Google Scholar] [CrossRef] [PubMed]
- Dudkowska, M.; Staniak, K.; Bojko, A.; Sikora, E. The role of autophagy in escaping therapy-induced polyploidy/senescence. Adv. Cancer Res. 2021, 150, 209–247. [Google Scholar] [PubMed]
- Shen, H.; Moran, D.M.; Maki, C.G. Transient nutlin-3a treatment promotes endoreduplication and the generation of therapy-resistant tetraploid cells. Cancer Res. 2008, 68, 8260–8268. [Google Scholar] [CrossRef] [Green Version]
- Glassmann, A.; Carrillo Garcia, C.; Janzen, V.; Kraus, D.; Veit, N.; Winter, J.; Probstmeier, R. Staurosporine induces the generation of polyploid giant cancer cells in non-small-cell lung carcinoma A549 cells. Anal. Cell. Pathol. (Amst.) 2018, 2018, 1754085. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, D.; Mandal, M. Senescence in polyploid giant cancer cells: A road that leads to chemoresistance. Cytokine Growth Factor Rev. 2020, 52, 68–75. [Google Scholar] [CrossRef]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discovery 2017, 3, 17049. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zhang, Q.; Wang, S.; Xie, S.; Fang, W.; Liu, Z.; Liu, J.; Yao, K. A fraction of CD133+ CNE2 cells is made of giant cancer cells with morphological evidence of asymmetric mitosis. J. Cancer 2015, 6, 1236–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy formation in doxorubicin-treated cancer cells can favor escape from senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D. Is senescence reversible? Curr. Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef]
- Czarnecka-Herok, J.; Sliwinska, M.A.; Herok, M.; Targonska, A.; Strzeszewska-Potyrala, A.; Bojko, A.; Wolny, A.; Mosieniak, G.; Sikora, E. Therapy-induced senescent/polyploid cancer cells undergo atypical divisions associated with altered expression of meiosis, spermatogenesis and EMT genes. Int. J. Mol. Sci. 2022, 23, 8288. [Google Scholar] [CrossRef]
- Chang, B.D.; Broude, E.V.; Fang, J.; Kalinichenko, T.V.; Abdryashitov, R.; Poole, J.C.; Roninson, I.B. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene 2000, 19, 2165–2170. [Google Scholar] [CrossRef] [Green Version]
- Xuan, B.; Ghosh, D.; Cheney, E.M.; Clifton, E.M.; Dawson, M.R. Dysregulation in actin cytoskeletal organization drives increased stiffness and migratory persistence in polyploidal giant cancer cells. Sci. Rep. 2018, 8, 11935. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Y.; Wu, M.; Liu, J.; Wu, H.; Xu, C.; Chen, L. Hypoxia-induced polypoid giant cancer cells in glioma promote the transformation of tumor-associated macrophages to a tumor-supportive phenotype. CNS Neurosci. Ther. 2022, 28, 1326–1338. [Google Scholar] [CrossRef]
- You, B.; Xia, T.; Gu, M.; Zhang, Z.; Zhang, Q.; Shen, J.; Fan, Y.; Yao, H.; Pan, S.; Lu, Y.; et al. AMPK-mTOR-mediated activation of autophagy promotes formation of dormant polyploid giant cancer cells. Cancer Res. 2022, 82, 846–858. [Google Scholar] [CrossRef]
- Bowers, R.R.; Andrade, M.F.; Jones, C.M.; White-Gilbertson, S.; Voelkel-Johnson, C.; Delaney, J.R. Autophagy modulating therapeutics inhibit ovarian cancer colony generation by polyploid giant cancer cells (PGCCs). BMC Cancer 2022, 22, 410. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Ali, A.M.; Murty, V.V.; Raza, A. Mutation in SF3B1 gene promotes formation of polyploid giant cells in Leukemia cells. Med. Oncol. 2022, 39, 65. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Guillon, J.; Ferbeyre, G. Senescence: A program in the road to cell elimination and cancer. Semin. Cancer Biol. 2022, 81, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Borkowska, A.; Olszewska, A.; Klemba, A.; Marciniak, M.; Synowiec, A.; Kieda, C. Polyploidy formation in cancer cells: How a Trojan horse is born. Semin. Cancer Biol. 2022, 81, 24–36. [Google Scholar] [CrossRef]
- Xuan, B.; Ghosh, D.; Dawson, M.R. Contributions of the distinct biophysical phenotype of polyploidal giant cancer cells to cancer progression. Semin. Cancer Biol. 2022, 81, 64–72. [Google Scholar] [CrossRef]
- Zhang, J.; Qiao, Q.; Xu, H.; Zhou, R.; Liu, X. Human cell polyploidization: The good and the evil. Semin. Cancer Biol. 2022, 81, 54–63. [Google Scholar] [CrossRef]
- Brown, J.M.; Wouters, B.G. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999, 59, 1391–1399. [Google Scholar]
- Beresford, M.J. Medical reductionism: Lessons from the great philosophers. Q. J. Med. 2010, 103, 721–724. [Google Scholar] [CrossRef]
- Kienle, G.; Kiene, H. From reductionism to holism: Systems-oriented approaches in cancer research. Glob. Adv. Health Med. 2012, 1, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Gilson, P.; Merlin, J.-L.; Harlé, A. Deciphering tumour heterogeneity: From tissue to liquid biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef] [PubMed]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; San Luis, B.; Lane, D.P. Intratumour heterogeneity of p53 expression; causes and consequences. J. Pathol. 2019, 249, 274–285. [Google Scholar] [CrossRef]
- Ramón y Cajal, S.; Sesé, M.; Capdevila, C.; Trond, A.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybinski, B.; Yun, K. Addressing intra-tumoral heterogeneity and therapy resistance. Oncotarget 2016, 7, 72322–72342. [Google Scholar] [CrossRef] [Green Version]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Rubinstein, L.V.; Shoemaker, R.H.; Paull, K.D.; Simon, R.M.; Tosini, S.; Skehan, P.; Scudiero, D.A.; Monks, A.; Boyd, M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990, 82, 1113–1118. [Google Scholar]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured tumor cell lines. J. Nat. Cancer Inst. 1991, 83, 757–766. [Google Scholar]
- Weinstein, J.N.; Myers, T.G.; O’Connor, P.M.; Friend, S.H.; Fornace, A.J.; Kohn, K.W.; Fojo, T.; Bates, S.E.; Rubinstein, L.V.; Anderson, N.L.; et al. An information-intensive approach to the molecular pharmacology of cancer. Science 1997, 275, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Leverkus, L. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot087379. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of cell viability by the MTT assay. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Kailen, W.G. Publish Houses of Brick, not Mansions of Straw. Nature 2017, 5454, 387. [Google Scholar]
- Kailen, W.G. Preclinical Cancer Target Validation: How Not to Be Wrong. NIH Wednesday Afternoon Lectures (WELS) Series. Available online: https://videocast.nih.gov/watch=27066 (accessed on 24 January 2018).
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef]
- Hilal, T.; Gonzalez-Velez, M.; Prasad, V. Limitations in clinical trials leading to anticancer drug approvals by the US Food and Drug Administration. JAMA Intern. Med. 2020, 180, 1108–1115. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. 2020, 19, 23–38. [Google Scholar] [CrossRef]
- Antolin, A.A.; Ameratunga, M.; Banerji, U.; Clarke, P.; Workman, P.; Al-Lazikani, B. The kinase polypharmacology landscape of clinical PARP Inhibitors. Sci. Rep. 2020, 10, 2585. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Messerschmidt, J.L.; Prendergast, G.C.; Messerschmidt, G.L. How cancers escape immune destruction and mechanisms of action for the new significantly active immune therapies: Helping nonimmunologists decipher recent advances. Oncologist 2016, 21, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calì, B.; Molon, B.; Viola, A. Tuning cancer fate: The unremitting role of host immunity. Open Biol. 2017, 7, 170006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Converg. Sci. Phys. Oncol. 2017, 3, 34002. [Google Scholar] [CrossRef] [PubMed]
- Borroni, E.M.; Grizzi, F. Cancer Immunoediting and beyond in 2021. Int. J. Mol. Sci. 2021, 22, 13275. [Google Scholar]
- Meng, S.; Tripathy, D.; Frenkel, E.P.; Shete, S.; Naftalis, E.Z.; Huth, J.F.; Beitsch, P.D.; Leitch, M.; Hoover, S.; Euhus, D.; et al. Circulating tumor cells in patients with breast cancer dormancy. Clin. Cancer Res. 2004, 10, 8152–8162. [Google Scholar] [CrossRef] [Green Version]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, L.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [PubMed] [Green Version]
- Candas-Green, D.; Xie, B.; Huang, J.; Fan, M.; Wang, A.; Menaa, C.; Zhang, Y.; Zhang, L.; Jing, D.; Azghadi, S.; et al. Dual blockade of CD47 and HER2 eliminates radioresistant breast cancer cells. Nat. Commun. 2020, 11, 4591. [Google Scholar]
- Upton, R.; Banuelos, A.; Feng, D.; Biswas, T.; Kao, K.; McKenna, K.; Willingham, S.; Ho, P.Y.; Rosental, B.; Tal, M.C.; et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc. Natl Acad. Sci. USA 2021, 118, e2026849118. [Google Scholar]
- Cao, X.; Li, B.; Chen, J.; Dang, J.; Chen, S.; Gunes, E.G.; Xu, B.; Tian, L.; Muend, S.; Raoof, M.; et al. Effect of cabazitaxel on macrophages improves CD47-targeted immunotherapy for triple-negative breast cancer. J. Immunother. Cancer 2021, 9, e002022. [Google Scholar]
- Holly, E. Top Journals Retract DNA-Repair Studies after Misconduct Probe: Investigation Found That Science and Nature Papers Contained Data Falsified by One Author. Nature News. 2019. Available online: https://www.nature.com/articles/d41586-019-00406-4 (accessed on 25 October 2022).
- Heng, J.; Heng, H.H. Genome chaos, information creation, and cancer emergence: Searching for new frameworks on the 50th anniversary of the “war on cancer”. Genes 2022, 13, 101. [Google Scholar] [CrossRef]
- Vainshelbaum, N.M.; Salmina, K.; Gerashchenko, B.I.; Lazovska, M.; Zayakin, P.; Cragg, M.S.; Pjanova, D.; Erenpreisa, J. Role of the circadian clock “death-loop” in the DNA damage response underpinning cancer treatment resistance. Cells 2022, 11, 880. [Google Scholar]
- Kasperski, A. Life entrapped in a network of atavistic attractors: How to find a rescue. J. Mol. Med. 2020, 23, 4017. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, F.; Xing, K.; He, X. The reverse evolution from multicellularity to unicellularity during carcinogenesis. Nat. Commun. 2015, 6, 6367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, M.; Wakasaki, T.; Toh, S. Stress-triggered atavistic reprogramming (STAR) addiction: Driving force behind head and neck cancer? Am. J. Cancer Res. 2016, 6, 1149–1166. [Google Scholar] [PubMed]
- Vincent, M. Resistance to cancer chemotherapy as an atavism? Bioessays 2016, 38, 1065. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [Green Version]
- Thomas, F.; Ujvari, B.; Renaud, F.; Vincent, M. Cancer adaptations: Atavism, de novo selection, or something in between? Bioessays 2017, 39, 1700039. [Google Scholar] [CrossRef]
- Chen, W.; Li, Y.; Wang, Z. Evolution of oncogenic signatures of mutation hotspots in tyrosine kinases supports the atavistic hypothesis of cancer. Sci. Rep. 2018, 8, 8256. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, V.F. Carcinogenesis: Recent insights in protist stem cell biology lead to a better understanding of atavistic mechanisms implied in cancer development. MOJ Tumor Res. 2018, 1, 18–29. [Google Scholar]
- Mazzocca, A. The systemic–evolutionary theory of the origin of cancer (SETOC): A new interpretative model of cancer as a complex biological system. Int. J. Mol. Sci. 2019, 20, 4885. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, V.F. aCLS cancers: Genomic and epigenetic changes transform the cell of origin of cancer into a tumorigenic pathogen of unicellular organization and lifestyle. Gene 2020, 726, 144174. [Google Scholar] [PubMed]
- Gorski, D. “Atavistic Oncology”: Another Dubious Cancer Therapy to be Avoided. Available online: https://sciencebasedmedicine.org/tag/atavism/ (accessed on 28 July 2014).
- Gorski, D. “Atavistic Oncology” Revisited: Dr. Frank Arguello Responds. Available online: https://sciencebasedmedicine.org/atavistic-oncology-revisited-dr-frank-arguello-responds/ (accessed on 18 August 2014).
- Lazarus-Barlow, W.S.; Leeming, J.H. The natural duration of cancer. Br. Med. J. 1924, 2, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, N.H.; Hulshof, M.C.; Bergman, J.J.; Geijsen, D.; Wilmink, J.W.; van Berge Henegouwen, M.I.; van Laarhoven, H.W. Acute toxicity of definitive chemoradiation in patients with inoperable or irresectable esophageal carcinoma. BMC Cancer 2014, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.R.; Mascitell, L. Surgery and cancer promotion: Are we trading beauty for cancer? Q. J. Med. 2011, 104, 811–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neeman, E.; Ben-Eliyahu, S. Surgery and stress promote cancer metastasis: New outlooks on perioperative mediating mechanisms and immune involvement. Brain Behav. Immun. 2013, 30, S32–S40. [Google Scholar] [CrossRef] [Green Version]
- Tohme, S.; Simmons, R.L.; Tsung, A. Surgery for cancer: A trigger for metastases. Cancer Res. 2017, 77, 1548–1552. [Google Scholar] [CrossRef] [Green Version]
- Krall, J.A.; Reinhardt, F.; Mercury, O.A.; Pattabiraman, D.R.; Brooks, M.W.; Dougan, M.; Lambert, A.W.; Bierie, B.; Ploegh, H.L.; Dougan, S.K.; et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 2018, 10, 436. [Google Scholar]
- Chen, Z.; Zhang, P.; Xu, Y.; Yan, J.; Liu, Z.; Lau, W.B.; Lau, B.; Li, Y.; Zhao, X.; Wei, Y.; et al. Surgical stress and cancer progression: The twisted tango. Mol. Cancer 2019, 18, 132. [Google Scholar]
- Mirzayans, R.; Andrais, B.; Murray, D. Viability assessment following anticancer treatment requires single-cell visualization. Cancers 2018, 10, 255. [Google Scholar]
- Mirzayans, R.; Andrais, B.; Murray, D. Do multiwell plate high throughput assays measure loss of cell viability following exposure to genotoxic agents? Int. J. Mol. Sci. 2017, 18, 1679. [Google Scholar] [CrossRef] [Green Version]
- Spiro, Z.; Kovacs, I.A.; Csermely, P. Drug-therapy networks and the prediction of novel drug targets. J. Biol. 2008, 7, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csermely, P.; Korcsmáros, T. Cancer-related networks: A help to understand, predict and change malignant transformation. Semin. Cancer Biol. 2013, 23, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Giuliani, A. Resolution of complex issues in genome regulation and cancer requires non-linear and network-based thermodynamics. Int. J. Mol. Sci. 2019, 21, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of cancer: Survival at the brink. Semin. Cancer Biol. 2022, 81, 119–131. [Google Scholar] [PubMed]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef]
- Rückert, M.; Flohr, A.S.; Hecht, M.; Gaipl, U.S. Radiotherapy and the immune system: More than just immune suppression. Stem Cells 2021, 39, 1155–1165. [Google Scholar] [CrossRef]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.X. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef]
- Brandmaier, A.; Formenti, S.C. The impact of radiation therapy on innate and adaptive tumor immunity. Semin. Radiat. Oncol. 2020, 30, 139–144. [Google Scholar] [CrossRef]
- Apetoh, L.; Ladoire, S.; Coukos, G.; Ghiringhelli, F. Combining immunotherapy and anticancer agents: The right path to achieve cancer cure? Ann. Oncol. 2015, 26, 1813–1823. [Google Scholar]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.B.; de Visser, K.E. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol. 2015, 36, 198–216. [Google Scholar] [CrossRef] [Green Version]
- de Biasi, A.R.; Villena-Vargas, J.; Adusumilli, P.S. Cisplatin-induced antitumor immunomodulation: A review of preclinical and clinical evidence. Clin. Cancer Res. 2014, 20, 5384–5391. [Google Scholar] [CrossRef] [Green Version]
- Fournier, C.; Rivera Vargas, T.; Martin, T.; Melis, A.; Apetoh, L. Immunotherapeutic properties of chemotherapy. Curr. Opin. Pharmacol. 2017, 35, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [Green Version]
- Hahn, T.; Polanczyk, M.J.; Borodovsky, A.; Ramanathapuram, L.V.; Akporiaye, E.T.; Ralph, S.J. Use of anti-cancer drugs, mitocans, to enhance the immune responses against tumors. Curr. Pharm. Biotechnol. 2013, 14, 357–376. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef]
- Zhang, J.; Pan, S.; Jian, C.; Hao, L.; Dong, J.; Sun, Q.; Jin, H.; Han, X. Immunostimulatory properties of chemotherapy in breast cancer: From immunogenic modulation mechanisms to clinical practice. Front. Immunol. 2022, 12, 819405. [Google Scholar] [CrossRef]
- Shurin, M.R.; Naiditch, H.; Gutkin, D.W.; Umansky, V.; Shurin, G.V. ChemoImmunoModulation: Immune regulation by the antineoplastic chemotherapeutic agents. Curr. Med. Chem. 2012, 19, 1792–1803. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Semenkow, S.; Li, S.; Kahlert, U.D.; Raabe, E.H.; Xu, J.; Arnold, A.; Janowski, M.; Oh, B.C.; Brandacher, G.; Bulte, L.W.M.; et al. An immunocompetent mouse model of human glioblastoma. Oncotarget 2017, 8, 61072–61082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basel, M.T.; Narayanan, S.; Ganta, C.; Shreshta, T.B.; Marquez, A.; Pyle, M.; Hill, J.; Bossmann, S.H.; Troyer, D.L. Developing a xenograft human tumor model in immunocompetent mice. Cancer Lett. 2018, 412, 256–263. [Google Scholar] [CrossRef]
- Wege, A.K. Humanized mouse models for the preclinical assessment of cancer immunotherapy. BioDrugs 2018, 32, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.C.; Aryee, K.E.; Cheng, M.; Kaur, P.; Keck, J.G.; Brehm, M.A. Creation of PDX-bearing humanized mice to study immuno-oncology. Methods Mol. Biol. 2019, 1953, 241–252. [Google Scholar]
- Available online: https://www.jax.org/news-and-insights/jax-blog/2020/july/why-humanized-mice (accessed on 25 October 2022).
- Coleman, M.P. War on cancer and the influence of the medical-industrial complex. J. Cancer Policy 2013, 1, e31–e34. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.T. The war on cancer: Are we winning? Tumor Biol. 2013, 34, 1275–1284. [Google Scholar] [CrossRef]
- Azpurua, J.; Seluanov, A. Long-lived cancer-resistant rodents as new model species for cancer research. Front. Genet. 2013, 3, 319. [Google Scholar] [CrossRef]
- Seluanov, A.; Gladyshev, V.N.; Vijg, J.; Gorbunova, V. Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer 2018, 18, 433–441. [Google Scholar] [CrossRef]
- Yamamura, Y.; Kawamura, Y.; Oka, K.; Miura, K. Carcinogenesis resistance in the longest-lived rodent, the naked mole-rat. Cancer Sci. 2022, in press. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirzayans, R.; Murray, D. What Are the Reasons for Continuing Failures in Cancer Therapy? Are Misleading/Inappropriate Preclinical Assays to Be Blamed? Might Some Modern Therapies Cause More Harm than Benefit? Int. J. Mol. Sci. 2022, 23, 13217. https://doi.org/10.3390/ijms232113217
Mirzayans R, Murray D. What Are the Reasons for Continuing Failures in Cancer Therapy? Are Misleading/Inappropriate Preclinical Assays to Be Blamed? Might Some Modern Therapies Cause More Harm than Benefit? International Journal of Molecular Sciences. 2022; 23(21):13217. https://doi.org/10.3390/ijms232113217
Chicago/Turabian StyleMirzayans, Razmik, and David Murray. 2022. "What Are the Reasons for Continuing Failures in Cancer Therapy? Are Misleading/Inappropriate Preclinical Assays to Be Blamed? Might Some Modern Therapies Cause More Harm than Benefit?" International Journal of Molecular Sciences 23, no. 21: 13217. https://doi.org/10.3390/ijms232113217