
4-Thiazolidinone-Bearing Hybrid Molecules in Anticancer Drug Design

 ,
,  , ,
, , 
{kind=link}
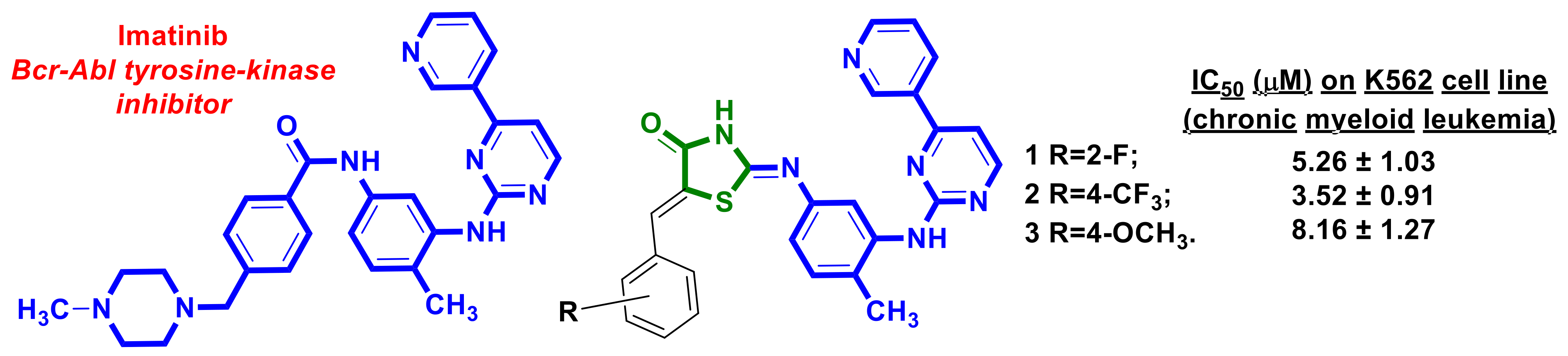{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
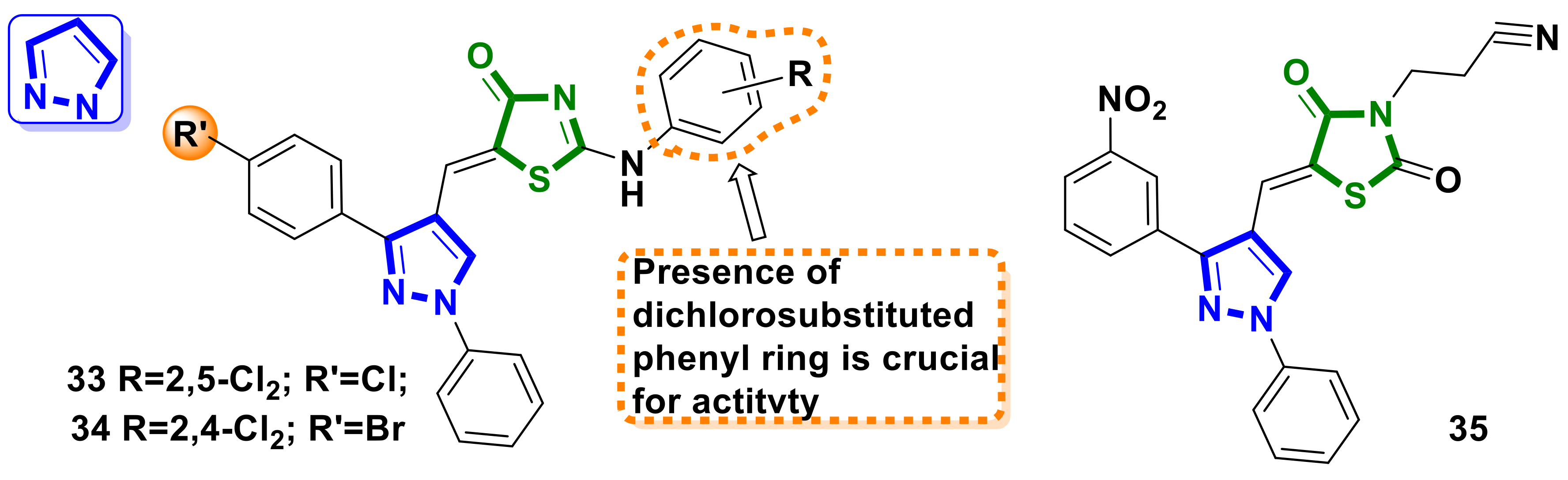{kind=link}
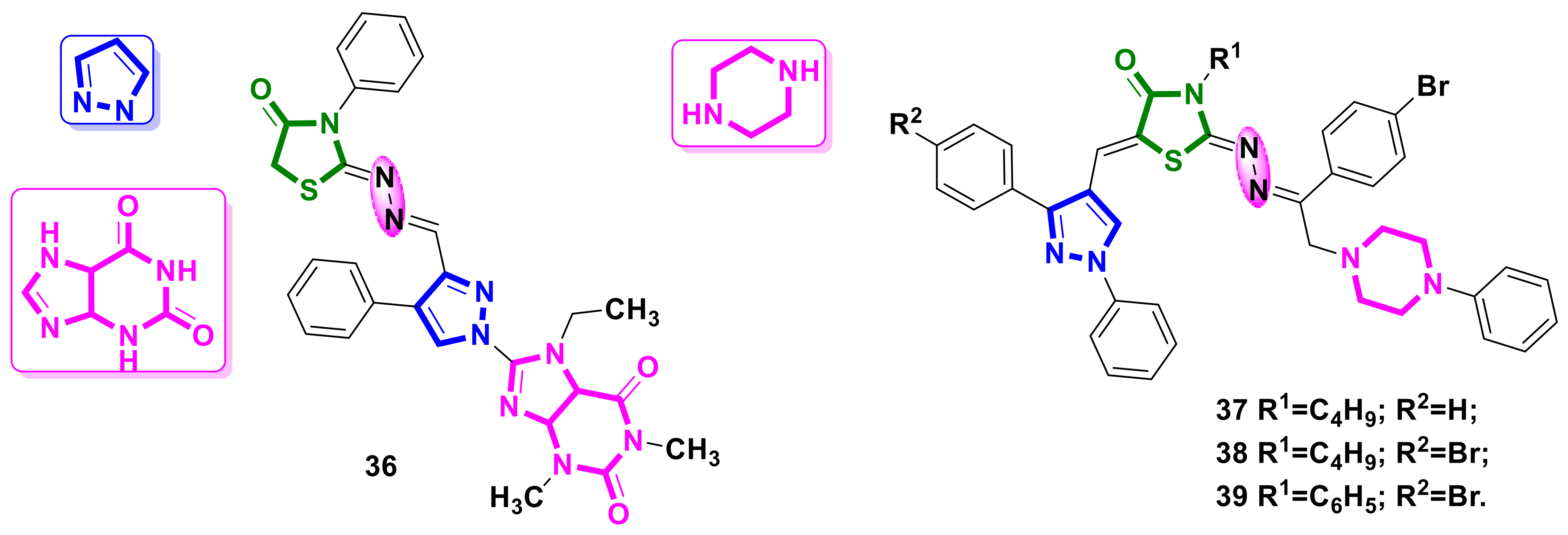{kind=link}
{kind=link}
{kind=link}
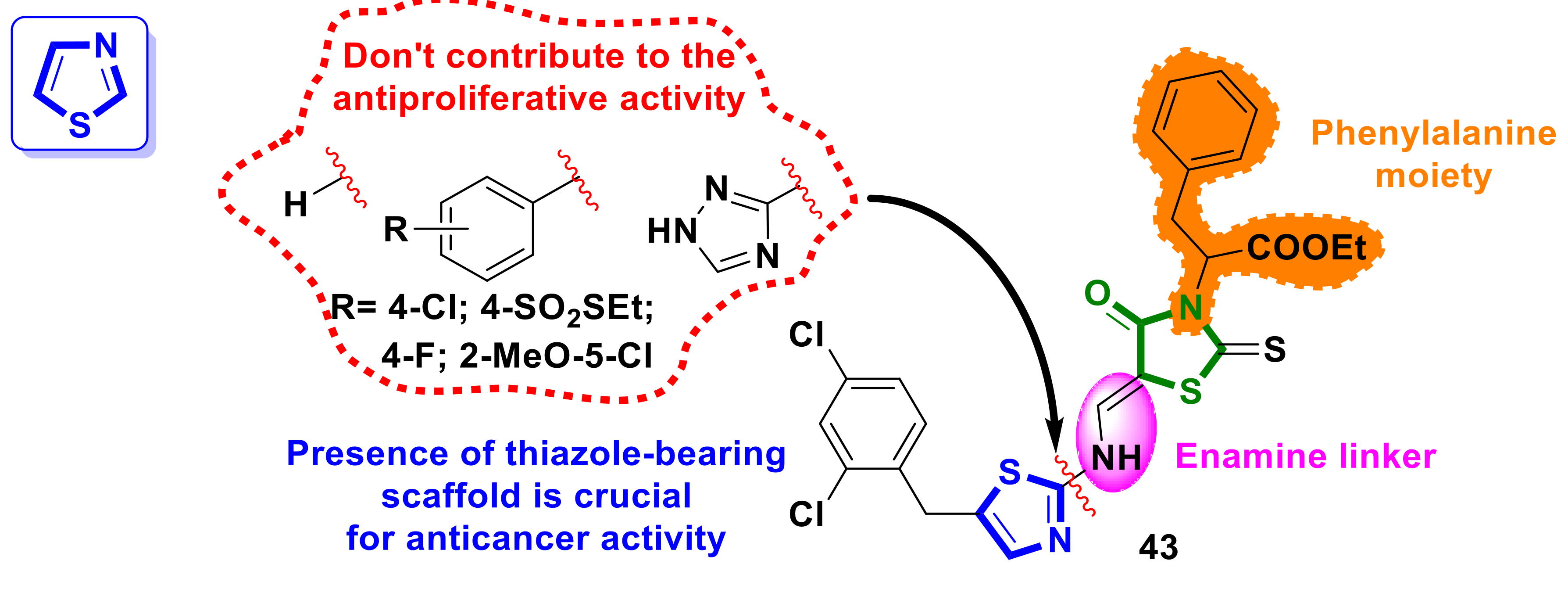{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Approved Drug–4-Thiazolidinones Hybrids
2.1. Anticancer Drug–4-Thiazolidinones Hybrids
2.2. NSAID–4-Thiazolidinones Hybrids
2.3. Antibacterial Drug–4-Thiazolidinones Hybrids
3. Natural Compound Scaffold–4-Thiazolidinones Hybrids
3.1. Monoterpene–4-Thiazolidinone Hybrids
3.2. Coumarin 4-Thiazolidinones Hybrids
3.3. Hybrids of 4-Thiazolidinones with Steroidal Skeleton
4. Privileged Heterocyclic Scaffolds–4-Thiazolidinones Hybrid Molecules
4.1. Furan–4-Thiazolidinone Hybrids
4.2. Pyrrole–4-Thiazolidinone Hybrids
4.3. Pyrazole–4-Thiazolidinone Hybrids
4.4. Pyrazole-Purine–4-Thiazolidinone Hybrids
4.5. Pyrazole-Piperazine–4-Thiazolidinone Hybrids
4.6. Pyrazoline-Pyrrole–4-Thiazolidinone Hybrids
4.7. Isoxazole–4-Thiazolidinone Hybrids
4.8. Thiazole–4-Thiazolidinone Hybrids
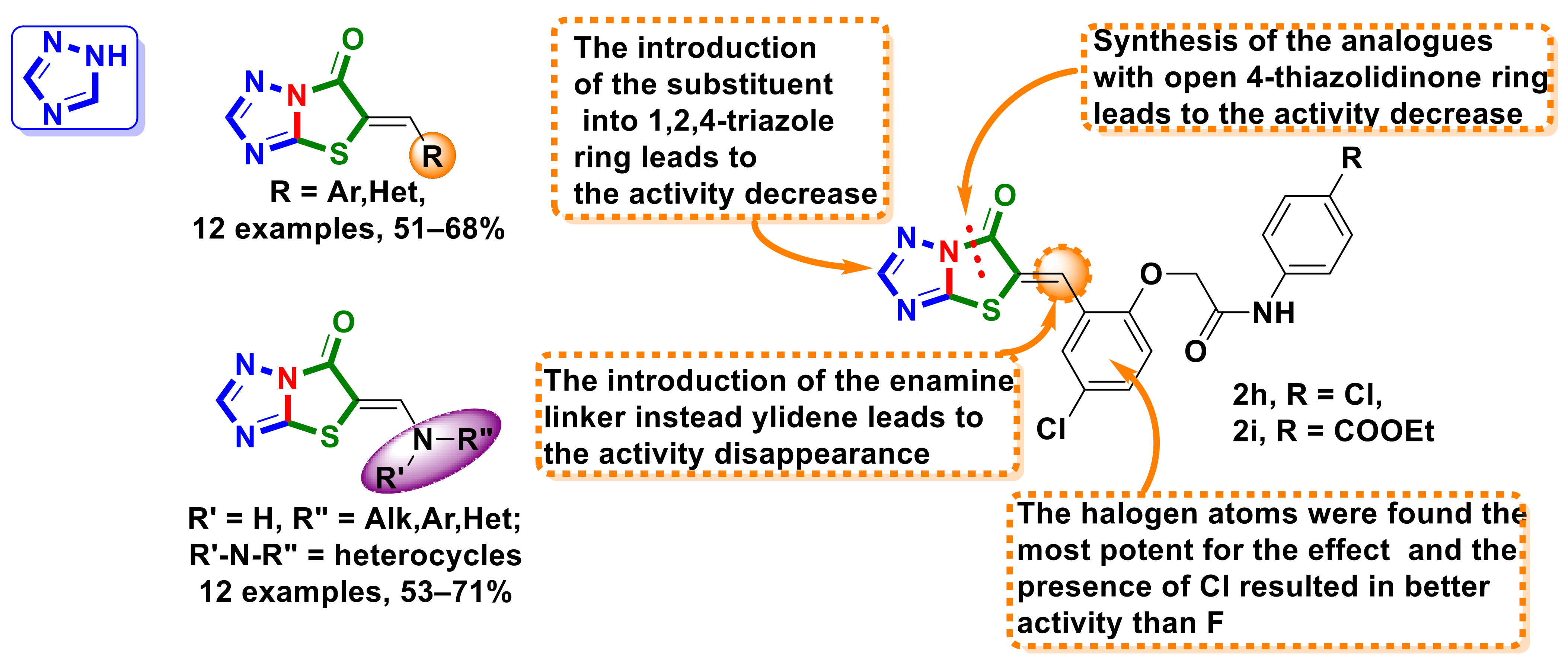
4.9. Triazole–4-Thiazolidinone Hybrids
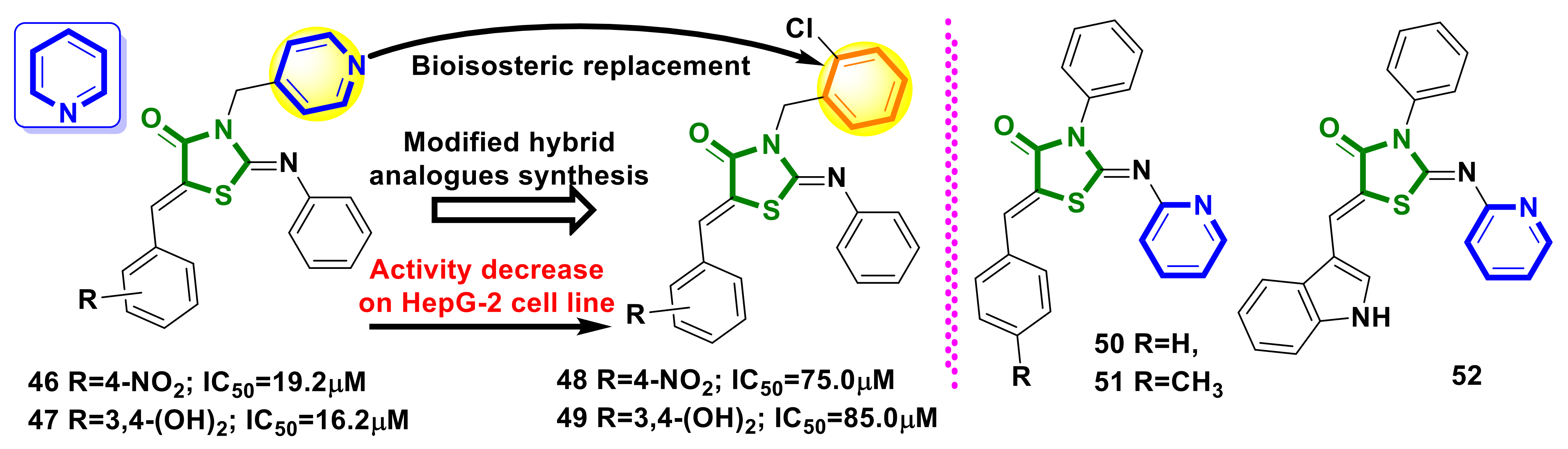
4.10. Pyridine–4-Thiazolidinone Hybrids
4.11. Pyridine-Piperazine–4-Thiazolidinone Hybrids
4.12. Pyridine-Thiazole–4-Thiazolidinone Hybrids
4.13. Indole–4-Thiazolidinone Hybrids
4.14. Isatine–4-Thiazolidinone Hybrids
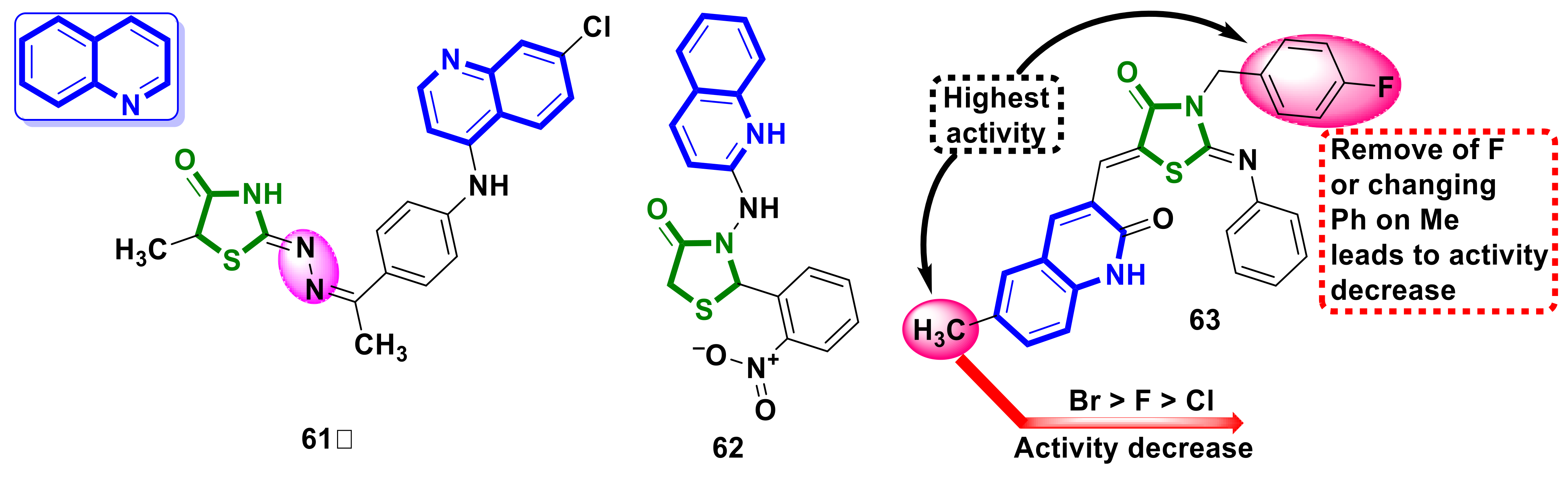
4.15. Quinoline–4-Thiazolidinone Hybrids
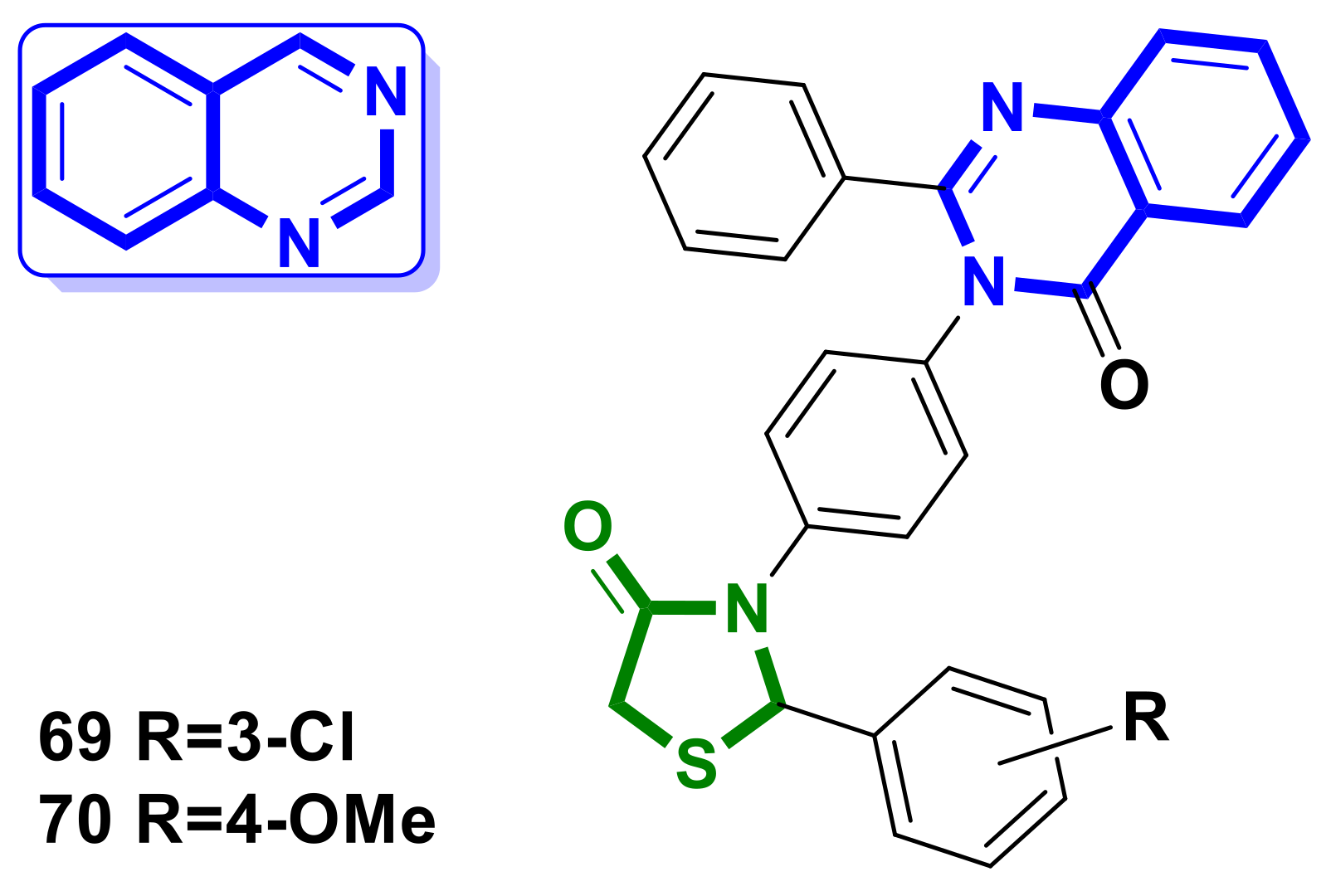
4.16. Quinazoline–4-Thiazolidinone Hybrids
4.17. Benzimidazole–4-Thiazolidinone Hybrids
4.18. Imidazopyridine–4-Thiazolidinone Hybrids
5. Empirical SAR, Mechanisms of Action, and Molecular Targets for 4-Thiazolidinone-Bearing Hybrid Molecules with Anticancer Activity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| CAIX | Carbonic anhydrase IX |
| CDC25 | Cell division cycle 25 phosphatases |
| CDK2 | Cyclin-dependent kinase 2 |
| CNS | Central nervous system |
| DNA | Deoxyribonucleic acid |
| DTP NCI | Developmental and Therapeutics Program at the National Cancer Institute |
| EGFR | Epidermal growth factor receptor |
| FDA | Food and Drug Administration |
| hCA | Human carbonic anhydrase |
| MMP-9 | Matrix metallopeptidase 9 |
| mRNA | Messenger ribonucleic acid |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| ROS | Reactive oxygen species |
| SAR | Structure–activity relationship |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kayl, A.E.; Meyers, C.A. Side-Effects of Chemotherapy and Quality of Life in Ovarian and Breast Cancer Patients. Curr. Opin. Obstet. Gynecol. 2006, 18, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Barreiro, E.J. Chapter 1. Privileged Scaffolds in Medicinal Chemistry: An Introduction; Royal Society of Chemistry: London, UK, 2015; pp. 1–15. [Google Scholar] [CrossRef]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An Aldose Reductase Inhibitor for the Treatment of Diabetic Neuropathy. Pharmacotherapy 2008, 28, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, H.E. Thiazolidinediones: The Forgotten Diabetes Medications. Curr. Diab. Rep. 2019, 19, 151. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, D.; Freedman, M.S.; Prinz, J. Ponesimod, a Selective S1P1 Receptor Modulator: A Potential Treatment for Multiple Sclerosis and Other Immune-Mediated Diseases. Ther. Adv. Chronic Dis. 2016, 7, 18–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, A. Ponesimod: First Approval. Drugs 2021, 81, 957–962. [Google Scholar] [CrossRef]
- Szczepański, J.; Tuszewska, H.; Trotsko, N. Anticancer Profile of Rhodanines: Structure–Activity Relationship (SAR) and Molecular Targets—A Review. Molecules 2022, 27, 3750. [Google Scholar] [CrossRef]
- Tilekar, K.; Shelke, O.; Upadhyay, N.; Lavecchia, A.; Ramaa, C.S. Current Status and Future Prospects of Molecular Hybrids with Thiazolidinedione (TZD) Scaffold in Anticancer Drug Discovery. J. Mol. Struct. 2022, 1250, 131767. [Google Scholar] [CrossRef]
- Türe, A.; Ergül, M.; Ergül, M.; Altun, A.; Küçükgüzel, İ. Design, Synthesis, and Anticancer Activity of Novel 4-Thiazolidinone-Phenylaminopyrimidine Hybrids. Mol. Divers. 2021, 25, 1025–1050. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Tiwari, A.K.; Sun, Y.; Ding, P.-R.; Ashby, C.R.; Chen, Z.-S. BCR-ABL Tyrosine Kinase Inhibitors in the Treatment of Philadelphia Chromosome Positive Chronic Myeloid Leukemia: A Review. Leuk. Res. 2010, 34, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-Inflammatory Drugs as Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadan, W.S.; Saleh, E.M.; Menon, V.; Vazhappilly, C.G.; Abdu-Allah, H.H.M.; El-Shorbagi, A.-N.A.; Mansour, W.; El-Awady, R. Induction of DNA Damage, Apoptosis and Cell Cycle Perturbation Mediate Cytotoxic Activity of New 5-Aminosalicylate–4-Thiazolinone Hybrid Derivatives. Biomed. Pharmacother. 2020, 131, 110571. [Google Scholar] [CrossRef]
- Shepeta, Y.; Lozynskyi, A.; Sulyma, M.; Nektegayev, I.; Grellier, P.; Lesyk, R. Synthesis and Biological Activity Evaluation of New Thiazolidinone-Diclofenac Hybrid Molecules. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 836–841. [Google Scholar] [CrossRef]
- Boyd, M.R. The NCI In Vitro Anticancer Drug Discovery Screen. In Anticancer Drug Development Guide; Humana Press: Totowa, NJ, USA, 1997; pp. 23–42. [Google Scholar]
- Shoemaker, R.H. The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Holbeck, S.L. Update on NCI in Vitro Drug Screen Utilities. Eur. J. Cancer 2004, 40, 785–793. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Collins, J.M.; Doroshow, J.H. Analysis of Food and Drug Administration–Approved Anticancer Agents in the NCI60 Panel of Human Tumor Cell Lines. Mol. Cancer Ther. 2010, 9, 1451–1460. [Google Scholar] [CrossRef] [Green Version]
- Holota, S.M.; Nektegayev, I.O.; Soronovych, I.I.; Chubuchna, I.I.; Kolishetska, M.A.; Sysak, S.P.; Regeda, M.S.; Lesyk, R.B. The Novel Pyrazolin-5-One Bearing Thiazolidin-4-Ones: Synthesis, Characterization and Biological Evaluation. Biopolym. Cell 2021, 37, 46–61. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; Gratteri, P.; Eissa, I.H.; Fares, M.; Ismael, O.E.; Ghabbour, H.A.; Elaasser, M.M.; Abdel-Aziz, H.A.; et al. Novel 4/3-((4-Oxo-5-(2-Oxoindolin-3-Ylidene)Thiazolidin-2-Ylidene)Amino) Benzenesulfonamides: Synthesis, Carbonic Anhydrase Inhibitory Activity, Anticancer Activity and Molecular Modelling Studies. Eur. J. Med. Chem. 2017, 139, 250–262. [Google Scholar] [CrossRef]
- Sunil Kumar, A.; Kudva, J.; Bharath, B.R.; Ananda, K.; Sadashiva, R.; Madan Kumar, S.; Revanasiddappa, B.C.; Kumar, V.; Rekha, P.D.; Naral, D. Synthesis, Structural, Biological and in Silico Studies of New 5-Arylidene-4-Thiazolidinone Derivatives as Possible Anticancer, Antimicrobial and Antitubercular Agents. New J. Chem. 2019, 43, 1597–1610. [Google Scholar] [CrossRef]
- Buzun, K.; Kryshchyshyn-Dylevych, A.; Senkiv, J.; Roman, O.; Gzella, A.; Bielawski, K.; Bielawska, A.; Lesyk, R. Synthesis and Anticancer Activity Evaluation of 5-[2-Chloro-3-(4-Nitrophenyl)-2-Propenylidene]-4-Thiazolidinones. Molecules 2021, 26, 3057. [Google Scholar] [CrossRef] [PubMed]
- Buzun, K.; Gornowicz, A.; Lesyk, R.; Kryshchyshyn-Dylevych, A.; Gzella, A.; Czarnomysy, R.; Latacz, G.; Olejarz-Maciej, A.; Handzlik, J.; Bielawski, K.; et al. 2-{5-[(Z,2Z)-2-Chloro-3-(4-Nitrophenyl)-2-Propenylidene]-4-Oxo-2-Thioxothiazolidin-3-Yl}-3-Methylbutanoic Acid as a Potential Anti-Breast Cancer Molecule. Int. J. Mol. Sci. 2022, 23, 4091. [Google Scholar] [CrossRef] [PubMed]
- Finiuk, N.; Kryshchyshyn-Dylevych, A.; Holota, S.; Klyuchivska, O.; Kozytskiy, A.; Karpenko, O.; Manko, N.; Ivasechko, I.; Stoika, R.; Lesyk, R. Novel Hybrid Pyrrolidinedione-Thiazolidinones as Potential Anticancer Agents: Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2022, 238, 114422. [Google Scholar] [CrossRef] [PubMed]
- Davison, E.K.; Brimble, M.A. Natural Product Derived Privileged Scaffolds in Drug Discovery. Curr. Opin. Chem. Biol. 2019, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sülsen, V.P.; Athanassopoulos, C.M.; Padrón, J.M.; Tamura, R.E. Editorial: Natural Compounds as Scaffolds for the Discovery of New Anti-Cancer Drugs: Focus on Terpenoids and Flavonoids. Front Pharmacol. 2022, 13, 984849. [Google Scholar] [CrossRef]
- Auhmani, A.; Fawzi, M.; Oubella, A.; el Mansouri, A.-E.; Bimoussa, A.; Ketatni, E.M.; Saadi, M.; el Ammari, L.; Itto, M.Y.A.; Morjani, H. Novel Hydrazono-2-iminothiazolidin-4-ones Based on a Monoterpenic Skelton as Potential Antitumor Agents: Synthesis, DFT Studies, in Vitro Cytotoxicity, Apoptosis Inducing Properties and Molecular Docking. Chem. Biodivers. 2022, 19, e202100836. [Google Scholar] [CrossRef]
- Oubella, A.; el Mansouri, A.E.; Fawzi, M.; Bimoussa, A.; Laamari, Y.; Auhmani, A.; Morjani, H.; Robert, A.; Riahi, A.; Youssef Ait Itto, M. Thiazolidinone-Linked1,2,3-Triazoles with Monoterpenic Skeleton as New Potential Anticancer Agents: Design, Synthesis and Molecular Docking Studies. Bioorg. Chem. 2021, 115, 105184. [Google Scholar] [CrossRef]
- El-Miligy, M.M.M.; Al-Kubeisi, A.K.; El-Zemity, S.R.; Nassra, R.A.; Abu-Serie, M.M.; Hazzaa, A.A. Discovery of Small Molecule Acting as Multitarget Inhibitor of Colorectal Cancer by Simultaneous Blocking of the Key COX-2, 5-LOX and PIM-1 Kinase Enzymes. Bioorg. Chem. 2021, 115, 105171. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Z. Coumarin-Containing Hybrids and Their Anticancer Activities. Eur. J. Med. Chem. 2019, 181, 111587. [Google Scholar] [CrossRef]
- Sigalapalli, D.K.; Kiranmai, G.; Tokala, R.; Tripura, C.; Ambatwar, R.; Nunewar, S.N.; Kadagathur, M.; Shankaraiah, N.; Nagesh, N.; Nagendra Babu, B.; et al. Targeting Tubulin Polymerization and DNA Binding of 4-Thiazolidinone–Umbelliferone Hybrids: Synthesis and Cytotoxicity Evaluation. New J. Chem. 2021, 45, 18908–18923. [Google Scholar] [CrossRef]
- Thacker, P.S.; Sridhar Goud, N.; Argulwar, O.S.; Soman, J.; Angeli, A.; Alvala, M.; Arifuddin, M.; Supuran, C.T. Synthesis and Biological Evaluation of Some Coumarin Hybrids as Selective Carbonic Anhydrase IX and XII Inhibitors. Bioorg. Chem. 2020, 104, 104272. [Google Scholar] [CrossRef] [PubMed]
- Sigalapalli, D.K.; Pooladanda, V.; Kadagathur, M.; Guggilapu, S.D.; Uppu, J.L.; Godugu, C.; Bathini, N.B.; Tangellamudi, N.D. Novel Chromenyl-Based 2-Iminothiazolidin-4-One Derivatives as Tubulin Polymerization Inhibitors: Design, Synthesis, Biological Evaluation and Molecular Modelling Studies. J. Mol. Struct. 2021, 1225, 128847. [Google Scholar] [CrossRef]
- Gupta, A.; Sathish Kumar, B.; Negi, A.S. Current Status on Development of Steroids as Anticancer Agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Živković, M.B.; Matić, I.Z.; Rodić, M.v.; Novaković, I.T.; Krivokuća, A.M.; Sladić, D.M.; Krstić, N.M. Anticancer Potential of New Steroidal Thiazolidin-4-One Derivatives. Mechanisms of Cytotoxic Action and Effects on Angiogenesis in Vitro. J. Steroid Biochem. Mol. Biol. 2017, 174, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Tahmasvand, R.; Bayat, P.; Vahdaniparast, S.M.; Dehghani, S.; Kooshafar, Z.; Khaleghi, S.; Almasirad, A.; Salimi, M. Design and Synthesis of Novel 4-Thiazolidinone Derivatives with Promising Anti-Breast Cancer Activity: Synthesis, Characterization, in Vitro and in Vivo Results. Bioorg. Chem. 2020, 104, 104276. [Google Scholar] [CrossRef]
- Shawky, A.M.; Abourehab, M.A.S.; Abdalla, A.N.; Gouda, A.M. Optimization of Pyrrolizine-Based Schiff Bases with 4-Thiazolidinone Motif: Design, Synthesis and Investigation of Cytotoxicity and Anti-Inflammatory Potency. Eur. J. Med. Chem. 2020, 185, 111780. [Google Scholar] [CrossRef]
- Bhat, M.; Poojary, B.; Kalal, B.S.; Gurubasavaraja Swamy, P.M.; Kabilan, S.; Kumar, V.; Shruthi, N.; Alias Anand, S.A.; Pai, V.R. Synthesis and Evaluation of Thiazolidinone–Pyrazole Conjugates as Anticancer and Antimicrobial Agents. Future Med. Chem. 2018, 10, 1017–1036. [Google Scholar] [CrossRef]
- Mushtaque, M.; Avecilla, F.; Haque, A.; Perwez, A.; Khan, M.S.; Rizvi, M.M.A. Experimental and Theoretical Studies of a Pyrazole-Thiazolidin-2,4-Di-One Hybrid. J. Mol. Struct. 2017, 1141, 417–427. [Google Scholar] [CrossRef]
- Afifi, O.S.; Shaaban, O.G.; Abd El Razik, H.A.; Shams El-Dine, S.E.D.A.; Ashour, F.A.; El-Tombary, A.A.; Abu-Serie, M.M. Synthesis and Biological Evaluation of Purine-Pyrazole Hybrids Incorporating Thiazole, Thiazolidinone or Rhodanine Moiety as 15-LOX Inhibitors Endowed with Anticancer and Antioxidant Potential. Bioorg. Chem. 2019, 87, 821–837. [Google Scholar] [CrossRef]
- El-Miligy, M.M.; Abd El Razik, H.A.; Abu-Serie, M.M. Synthesis of Piperazine-Based Thiazolidinones as VEGFR2 Tyrosine Kinase Inhibitors Inducing Apoptosis. Future Med. Chem. 2017, 9, 1709–1729. [Google Scholar] [CrossRef] [PubMed]
- Elewa, S.I.; El-Farargy, A.F.; Nafie, M.S.; Mansour, E. Synthesis, and Cytotoxic Activity of Novel Pyrazoline-Thiazolidinone Derivatives with Molecular Docking Studies. Polycycl. Aromat. Compd. 2022, 42, 1–19. [Google Scholar] [CrossRef]
- Ramesh, G.; Rathnakar, B.; Narsaiah, C.; Rameshwar, N.; Srinivas, M.; Namratha, V.; Durgaiah, G.; Reddy, Y.N.; Reddy, B.V.; Satyanarayana, M. Synthesis, DFT Computations, Molecular Docking Studies and Anticancer Activity of 2-(4-Fluorophenyl)-3-(5-Methylisoxazol-3-Yl)Thiazolidin-4-One. Chem. Data Collect. 2022, 39, 100859. [Google Scholar] [CrossRef]
- Holota, S.; Kryshchyshyn, A.; Derkach, H.; Trufin, Y.; Demchuk, I.; Gzella, A.; Grellier, P.; Lesyk, R. Synthesis of 5-Enamine-4-Thiazolidinone Derivatives with Trypanocidal and Anticancer Activity. Bioorg. Chem. 2019, 86, 126–136. [Google Scholar] [CrossRef]
- Holota, S.; Komykhov, S.; Sysak, S.; Gzella, A.; Cherkas, A.; Lesyk, R. Synthesis, Characterization and In Vitro Evaluation of Novel 5-Ene-Thiazolo[3,2-b][1,2,4]Triazole-6(5H)-Ones as Possible Anticancer Agents. Molecules 2021, 26, 1162. [Google Scholar] [CrossRef]
- Ansari, M.F.; Idrees, D.; Hassan, M.I.; Ahmad, K.; Avecilla, F.; Azam, A. Design, Synthesis and Biological Evaluation of Novel Pyridine-Thiazolidinone Derivatives as Anticancer Agents: Targeting Human Carbonic Anhydrase IX. Eur. J. Med. Chem. 2018, 144, 544–556. [Google Scholar] [CrossRef]
- Mushtaque, M.; Avecilla, F.; Hafeez, Z.b.; Rizvi, M.M.A. Synthesis, Characterization, Molecular Docking, and Anticancer Evaluation of 4-Thiazolidinone Analogues. J. Heterocycl. Chem. 2019, 56, 1794–1805. [Google Scholar] [CrossRef]
- Campos, J.C.; Campos, P.T.; Bona, N.P.; Soares, M.S.; Souza, P.O.; Braganhol, E.; Cunico, W.; Siqueira, G.M. Synthesis and Biological Evaluation of Novel 2-Imino-4-Thiazolidinones as Potential Antitumor Agents for Glioblastoma. Med. Chem. 2022, 18, 452–462. [Google Scholar] [CrossRef]
- Demirci, S.; Hayal, T.B.; Kıratlı, B.; Şişli, H.B.; Demirci, S.; Şahin, F.; Doğan, A. Design and Synthesis of Phenylpiperazine Derivatives as Potent Anticancer Agents for Prostate Cancer. Chem. Biol. Drug Des. 2019, 94, 1584–1595. [Google Scholar] [CrossRef]
- Hussein, E.M.; Alsantali, R.I.; Morad, M.; Obaid, R.J.; Altass, H.M.; Sayqal, A.; Abourehab, M.A.S.; Elkhawaga, A.A.; Aboraia, A.S.M.; Ahmed, S.A. Bioactive Fluorenes. Part III: 2,7-Dichloro-9H-Fluorene-Based Thiazolidinone and Azetidinone Analogues as Anticancer and Antimicrobial against Multidrug Resistant Strains Agents. BMC Chem. 2020, 14, 1–24. [Google Scholar] [CrossRef]
- Sigalapalli, D.K.; Pooladanda, V.; Singh, P.; Kadagathur, M.; Guggilapu, S.D.; Uppu, J.L.; Tangellamudi, N.D.; Gangireddy, P.K.; Godugu, C.; Bathini, N.B. Discovery of Certain Benzyl/Phenethyl Thiazolidinone-Indole Hybrids as Potential Anti-Proliferative Agents: Synthesis, Molecular Modeling and Tubulin Polymerization Inhibition Study. Bioorg. Chem. 2019, 92, 103188. [Google Scholar] [CrossRef]
- De Oliveira, J.F.; Lima, T.S.; Vendramini-Costa, D.B.; de Lacerda Pedrosa, S.C.B.; Lafayette, E.A.; da Silva, R.M.F.; de Almeida, S.M.V.; de Moura, R.O.; Ruiz, A.L.T.G.; de Carvalho, J.E.; et al. Thiosemicarbazones and 4-Thiazolidinones Indole-Based Derivatives: Synthesis, Evaluation of Antiproliferative Activity, Cell Death Mechanisms and Topoisomerase Inhibition Assay. Eur. J. Med. Chem. 2017, 136, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Kryshchyshyn-Dylevych, A.; Radko, L.; Finiuk, N.; Garazd, M.; Kashchak, N.; Posyniak, A.; Niemczuk, K.; Stoika, R.; Lesyk, R. Synthesis of Novel Indole-Thiazolidinone Hybrid Structures as Promising Scaffold with Anticancer Potential. Bioorg. Med. Chem. 2021, 50, 116453. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D.; Keating, G.M. Sunitinib. Drugs 2006, 66, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, M.; Eldehna, W.M.; Almahli, H.; Elgez, A.; Fares, M.; Elaasser, M.M.; Abdel-Aziz, H.A. Novel Thiazolidinone/Thiazolo[3,2-a] Benzimidazolone-Isatin Conjugates as Apoptotic Anti-Proliferative Agents towards Breast Cancer: One-Pot Synthesis and in Vitro Biological Evaluation. Molecules 2018, 23, 1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, M.A.; Zaki, M.Y.; Lotfy, R.A.; Mahmoud, W.R. Insight on a New Indolinone Derivative as an Orally Bioavailable Lead Compound against Renal Cell Carcinoma. Bioorg. Chem. 2021, 112, 104985. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Leja, M.L.; Kaminskyy, D.v.; Binduga, U.E.; Pinyazhko, O.R.; Lesyk, R.B.; Gmiński, J. Study of Novel Anticancer 4-Thiazolidinone Derivatives. Chem. Biol. Interact. 2017, 262, 46–56. [Google Scholar] [CrossRef]
- Batran, R.Z.; El-Daly, S.M.; El-Kashak, W.A.; Ahmed, E.Y. Design, Synthesis, and Molecular Modeling of Quinoline-Based Derivatives as Anti-Breast Cancer Agents Targeting EGFR/AKT Signaling Pathway. Chem. Biol. Drug Des. 2022, 99, 470–482. [Google Scholar] [CrossRef]
- Nafie, M.S.; Kishk, S.M.; Mahgoub, S.; Amer, A.M. Quinoline-Based Thiazolidinone Derivatives as Potent Cytotoxic and Apoptosis-Inducing Agents through EGFR Inhibition. Chem. Biol. Drug Des. 2022, 99, 547–560. [Google Scholar] [CrossRef]
- Kumar, V.; Rai, V.M.; Udupi, V.; Shivalingegowda, N.; Pai, V.R.; Krishnappagowda, L.N.; Poojary, B. Synthesis, Crystal Structure, Anticancer and Molecular Docking Studies of Quinolinone-Thiazolidinone Hybrid Molecules. J. Iran. Chem. Soc. 2022, 19, 793–808. [Google Scholar] [CrossRef]
- Qi, B.; Yang, Y.; He, H.; Yue, X.; Zhou, Y.; Zhou, X.; Chen, Y.; Liu, M.; Zhang, A.; Wei, F. Identification of Novel N-(2-Aryl-1, 3-Thiazolidin-4-One)-N-Aryl Ureas Showing Potent Multi-Tyrosine Kinase Inhibitory Activities. Eur. J. Med. Chem. 2018, 146, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Yang, Y.; Gong, G.; He, H.; Yue, X.; Xu, X.; Hu, Y.; Li, J.; Chen, T.; Wan, X.; et al. Discovery of N-(4-((7-(3-(4-Ethylpiperazin-1-yl)Propoxy)-6-Methoxyquinolin-4-yl)Oxy)-3,5-Difluorophenyl)-N-(2-(2,6-Difluorophenyl)-4-Oxothiazolidin-3-yl)Urea as a Multi-Tyrosine Kinase Inhibitor for Drug-Sensitive and Drug-Resistant Cancers Treatment. Eur. J. Med. Chem. 2019, 163, 10–27. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, X.; Wang, F.; He, H.; Gong, G.; Xiong, L.; Qi, B. Identification of Novel Quinoline Analogues Bearing Thiazolidinones as Potent Kinase Inhibitors for the Treatment of Colorectal Cancer. Eur. J. Med. Chem. 2020, 204, 112643. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, X.; Wang, F.; He, H.; Qi, B. Discovery of 4-((4-(4-(3-(2-(2,6-Difluorophenyl)-4-Oxothiazolidin-3-yl)Ureido)-2-Fluorophenoxy)-6-Methoxyquinolin-7-yl)Oxy)-N,N-Diethylpiperidine-1-Carboxamide as Kinase Inhibitor for the Treatment of Colorectal Cancer. Bioorg. Chem. 2021, 106, 104511. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Wang, F.; He, H.; Fan, M.; Hu, L.; Xiong, L.; Gong, G.; Shi, S.; Song, X. Identification of (S)-1-(2-(2,4-Difluorophenyl)-4-Oxothiazolidin-3-Yl)-3-(4-((7-(3-(4-Ethylpiperazin-1-Yl)Propoxy)-6-Methoxyquinolin-4-Yl)Oxy)-3,5-Difluorophenyl)Urea as a Potential Anti-Colorectal Cancer Agent. Eur. J. Med. Chem. 2022, 239, 114561. [Google Scholar] [CrossRef]
- Thakral, S.; Saini, D.; Kumar, A.; Jain, N.; Jain, S. A Synthetic Approach and Molecular Docking Study of Hybrids of Quinazolin-4-Ones and Thiazolidin-4-Ones as Anticancer Agents. Med. Chem. Res. 2017, 26, 1595–1604. [Google Scholar] [CrossRef]
- Sharma, P.; Reddy, T.S.; Kumar, N.P.; Senwar, K.R.; Bhargava, S.K.; Shankaraiah, N. Conventional and Microwave-Assisted Synthesis of New 1H-Benzimidazole-Thiazolidinedione Derivatives: A Potential Anticancer Scaffold. Eur. J. Med. Chem. 2017, 138, 234–245. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Husain, A.; Alam, O.; Khan, S.A.; Ahmad, A.; Haider, M.R.; Alam, M.A. Design, Synthesis, and Biological Evaluation of Imidazopyridine-linked Thiazolidinone as Potential Anticancer Agents. Arch. Pharm. 2020, 353, 2000071. [Google Scholar] [CrossRef]






























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roszczenko, P.; Holota, S.; Szewczyk, O.K.; Dudchak, R.; Bielawski, K.; Bielawska, A.; Lesyk, R. 4-Thiazolidinone-Bearing Hybrid Molecules in Anticancer Drug Design. Int. J. Mol. Sci. 2022, 23, 13135. https://doi.org/10.3390/ijms232113135
Roszczenko P, Holota S, Szewczyk OK, Dudchak R, Bielawski K, Bielawska A, Lesyk R. 4-Thiazolidinone-Bearing Hybrid Molecules in Anticancer Drug Design. International Journal of Molecular Sciences. 2022; 23(21):13135. https://doi.org/10.3390/ijms232113135
Chicago/Turabian StyleRoszczenko, Piotr, Serhii Holota, Olga Klaudia Szewczyk, Rostyslav Dudchak, Krzysztof Bielawski, Anna Bielawska, and Roman Lesyk. 2022. "4-Thiazolidinone-Bearing Hybrid Molecules in Anticancer Drug Design" International Journal of Molecular Sciences 23, no. 21: 13135. https://doi.org/10.3390/ijms232113135