
Alzheimer’s Disease: From Immune Homeostasis to Neuroinflammatory Condition
 ,
,  ,
,
Abstract
:1. Introduction
2. Neuropathological Hallmarks of Alzheimer’s Disease
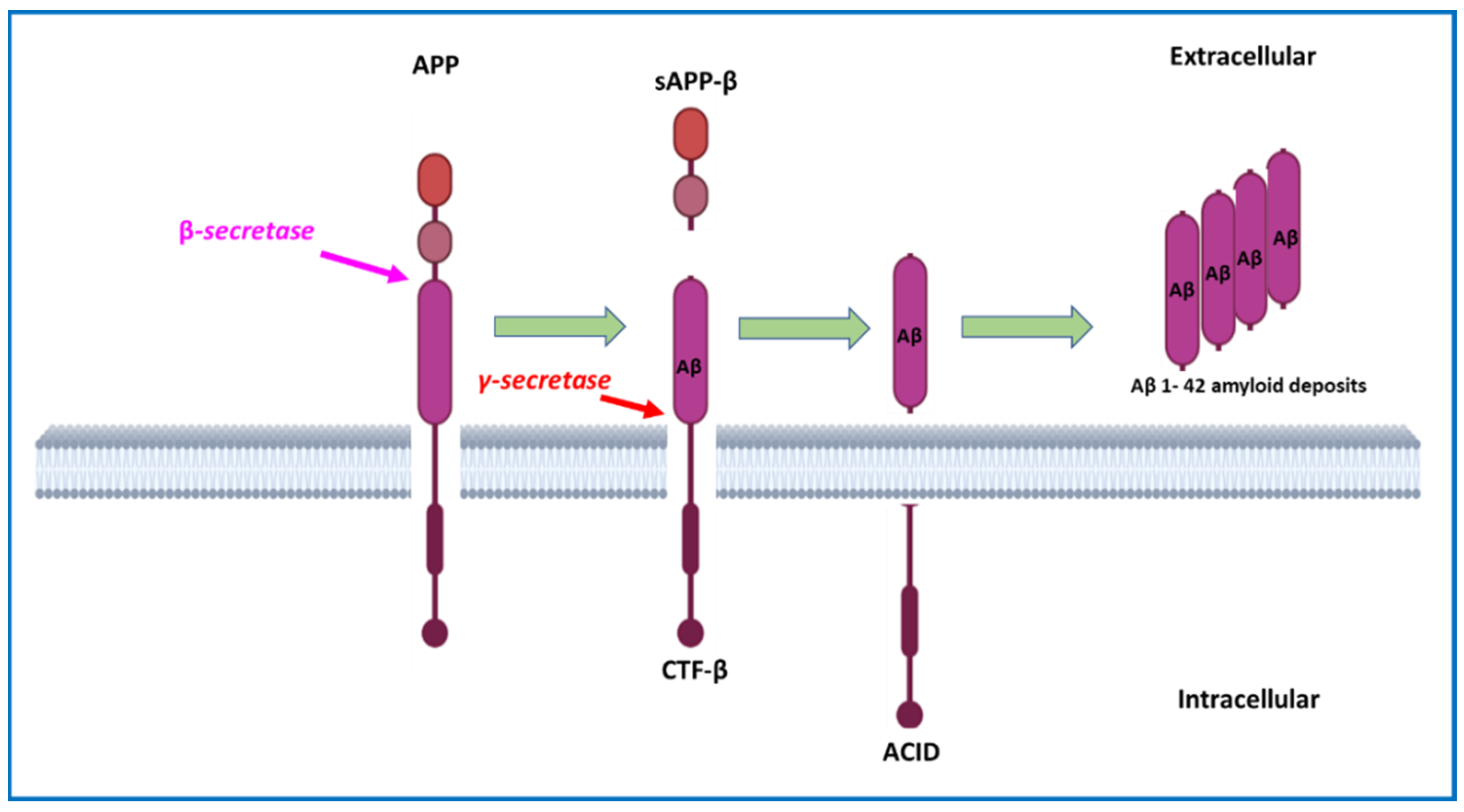
2.1. Amyloid Plaques and Amyloid Cascade Hypothesis
2.2. Neurofibrillary Tangles
2.3. Alzheimer’s Disease Pathogenesis Theories
- -
- First, cholinergic theory, which suggests that AD is caused by a degenerative process that is capable of selectively damaging groups of cholinergic neurons in the hippocampus, frontal cortex, amygdala, nucleus basalis, and medial septum, regions and structures that serve important functional roles in attention, learning, and memory. This selective alteration leads to the reduction of cholinergic markers such as acetylcholinesterase [44]
- -
- -
- Third, tauogenic theory, which proposes that tau protein aggregation and, consequently, NFT development, directly cause neuronal abnormalities, activating a neuroinflammatory condition in the extracellular space and inducing neuronal apoptosis [46].
3. The Role of CNS Immune System
3.1. Physiological Role of CNS Innate Immune Cells
3.2. Role of CNS Innate Immune System in Alzheimer’s Disease
3.2.1. Microglia and Aging in Alzheimer’s Disease
3.2.2. Microglia and Aβ-Protein in Alzheimer’s Disease
3.3. Physiological Role of Peripheral Adaptive Immune Cells
3.4. Role of Peripheral Adaptive Immune System in Alzheimer’s Disease

{kind=link}
{kind=link}
| Tissue | Species | Increased CD4+ | Increased Th17 | Reduced Treg | Increased CD8+ | Ref |
|---|---|---|---|---|---|---|
| AD Postmortem brain tissue | human | yes | no | no | yes | [149] |
| AD Postmortem brain tissue | human | yes | no | no | yes | [151] |
| AD Postmortem brain tissue | human | yes | no | no | yes | [152] |
| AD Postmortem brain tissue | human | yes | no | no | yes | [150] |
| AD Postmortem brain tissue | human | yes | no | no | no | [136] |
| Brain tissue and peripheral blood | transgenic APP mouse | yes | no | no | no | [145] |
| Brain tissue/peripheral blood | transgenic APP rat/human | yes | no | no | no | [146] |
| Peripheral blood | human | no | no | yes | no | [155] |
| Brain tissue | transgenic APP mouse | yes | no | no | no | [144] |
| Peripheral blood | human | no | yes | no | no | [160] |
| Brain tissue | Mouse | no | yes | no | no | [163] |
| Brain tissue | 5xFAD AD mouse | no | no | no | no | [170] |
| AD Postmortem brain tissue | human | yes | no | no | yes | [148] |
| Brain tissue | transgenic APP mouse | yes | no | no | no | [147] |
| Brain tissue | 3XTg AD mouse | no | no | yes | no | [166] |
| Brain tissue/peripheral blood | transgenic APP1 mouse | no | no | yes | no | [167] |
| Brain tissue | mammalian | yes | no | no | no | [137] |
| Peripheral blood | human | no | no | no | no | [165] |
| Peripheral blood | human | no | yes | yes | yes | [161] |
| Peripheral blood | human | no | yes | yes | yes | [90] |
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2017 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Lovell, M.A. Four-Hydroxynonenal, a Product of Lipid Peroxidation, Is Increased in the Brain in Alzheimer’s Disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Harrington, W.W.; Britt, C.S.; Wilson, J.G.; Milliken, N.O.; Binz, J.G.; Lobe, D.C.; Oliver, W.R.; Lewis, M.C.; Ignar, D.M. The Effect of PPARα, PPARδ, PPARγ, and PPARpan Agonists on Body Weight, Body Mass, and Serum Lipid Profiles in Diet-Induced Obese AKR/J Mice. PPAR Res. 2007, 2007, 097125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabert, M.H.; Liu, X.; Doty, R.L.; Serby, M.; Zamora, D.; Pelton, G.H.; Marder, K.; Albers, M.W.; Stern, Y.; Devanand, D.P. A 10-Item Smell Identification Scale Related to Risk for Alzheimer’s Disease. Ann. Neurol. 2005, 58, 155–160. [Google Scholar] [CrossRef]
- Castellani, R.J.; Perry, G. Pathogenesis and Disease-Modifying Therapy in Alzheimer’s Disease: The Flat Line of Progress. Arch. Med. Res. 2012, 43, 694–698. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Ankarcrona, M.; Mangialasche, F.; Winblad, B. Rethinking Alzheimer’s Disease Therapy: Are Mitochondria the Key? J. Alzheimer’s Dis. 2010, 20, S579–S590. [Google Scholar] [CrossRef] [Green Version]
- Davies, P. Selective Loss of Central Cholinergic Neurons in Alzheimer’s Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Lee Mosley, R.; Benner, E.J.; Kadiu, I.; Thomas, M.; Boska, M.D.; Hasan, K.; Laurie, C.; Gendelman, H.E. Neuroinflammation, Oxidative Stress, and the Pathogenesis of Parkinson’s Disease. Clin. Neurosci. Res. 2006, 6, 261–281. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; de Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, E.D.; Mucke, L. 100 Years and Counting: Prospects for Defeating Alzheimer’s Disease. Science 2006, 314, 781–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer’s Disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Schellenberg, G.D.; Bird, T.D.; Wijsman, E.M.; Orr, H.T.; Anderson, L.; Nemens, E.; White, J.A.; Bonnycastle, L.; Weber, J.L.; Alonso, M.E.; et al. Genetic Linkage Evidence for a Familial Alzheimer’s Disease Locus on Chromosome 14. Science 1992, 258, 668–671. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; van Eldik, L.J. Using Mice to Model Alzheimer’s Dementia: An Overview of the Clinical Disease and the Preclinical Behavioral Changes in 10 Mouse Models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R. The Development of Amyloid Beta Protein Deposits in the Aged Brain. Sci. Aging Knowl. Environ. 2006, 2006, re1. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A. A Review on Alzheimer’s Disease Pathophysiology and Its Management: An Update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of γ-Secretase Activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef]
- Goedert, M. Oskar Fischer and the Study of Dementia. Brain 2008, 132, 1102–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two Transmembrane Aspartates in Presenilin-1 Required for Presenilin Endoproteolysis and γ-Secretase Activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Cheung, T.T.; Cai, X.-D.; Odaka, A.; Otvos, L.; Eckman, C.; Golde, T.E.; Younkin, S.G. An Increased Percentage of Long Amyloid β Protein Secreted by Familial Amyloid β Protein Precursor (ΒApp717) Mutants. Science 1994, 264, 1336–1340. [Google Scholar] [CrossRef]
- Cras, P.; Kawai, M.; Lowery, D.; Gonzalez-DeWhitt, P.; Greenberg, B.; Perry, G. Senile Plaque Neurites in Alzheimer Disease Accumulate Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 1991, 88, 7552–7556. [Google Scholar] [CrossRef] [Green Version]
- Gotz, J.; Chen, F.; van Dorpe, J.; Nitsch, R.M. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Aβ42 Fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.-L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.-H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Quintero-Monzon, O.; Ostaszewski, B.L.; Podlisny, D.R.; Cavanaugh, W.T.; Yang, T.; Holtzman, D.M.; Cirrito, J.R.; Selkoe, D.J. Dynamic Analysis of Amyloid β-Protein in Behaving Mice Reveals Opposing Changes in ISF versus Parenchymal Aβ during Age-Related Plaque Formation. J. Neurosci. 2011, 31, 15861–15869. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of Abnormally Phosphorylated Tau in the Breakdown of Microtubules in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, D.P. Neuropathology of Alzheimer’s Disease. Mt. Sinai J. Med. J. Transl. Pers. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef]
- Dickson, D.W.; Crystal, H.A.; Mattiace, L.A.; Masur, D.M.; Blau, A.D.; Davies, P.; Yen, S.-H.; Aronson, M.K. Identification of Normal and Pathological Aging in Prospectively Studied Nondemented Elderly Humans. Neurobiol. Aging 1992, 13, 179–189. [Google Scholar] [CrossRef]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical Neurofibrillary Tangles Correlate with Dementia Severity in Alzheimer’s Disease. Arch. Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Saito, T.; Saido, T.C. Neuroinflammation in Mouse Models of Alzheimer’s Disease. Clin. Exp. Neuroimmunol. 2018, 9, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.A. Risk Factors for Alzheimer’s Disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia Prevention, Intervention, and Care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef] [Green Version]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic Activity in Alzheimer’s Disease: Causes and Clinical Relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, W.-J.; Chen, W.-W. Microwaves and Alzheimer’s Disease. Exp. Ther. Med. 2016, 12, 1969–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumtaz, S.; Rana, J.N.; Choi, E.H.; Han, I. Microwave Radiation and the Brain: Mechanisms, Current Status, and Future Prospects. Int. J. Mol. Sci. 2022, 23, 9288. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The Cholinergic Hypothesis of Alzheimer’s Disease: A Review of Progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R. The Amyloid Hypothesis: Let Sleeping Dogmas Lie? Neurobiol. Aging 2002, 23, 1101–1105. [Google Scholar] [CrossRef]
- Mudher, A.; Lovestone, S. Alzheimer’s Disease—Do Tauists and Baptists Finally Shake Hands? Trends Neurosci. 2002, 25, 22–26. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [Green Version]
- Hensley, K. Neuroinflammation in Alzheimer’s Disease: Mechanisms, Pathologic Consequences, and Potential for Therapeutic Manipulation. J. Alzheimer’s Dis. 2010, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting Neuroinflammation in Alzheimer’s Disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyzériat, K.; Zilli, T.; Millet, P.; Frisoni, G.B.; Garibotto, V.; Tournier, B.B. Learning from the Past: A Review of Clinical Trials Targeting Amyloid, Tau and Neuroinflammation in Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 112–125. [Google Scholar] [CrossRef]
- Nazem, A.; Mansoori, G.A. Nanotechnology Solutions for Alzheimer’s Disease: Advances in Research Tools, Diagnostic Methods and Therapeutic Agents. J. Alzheimer’s Dis. 2008, 13, 199–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikelenboom, P.; van Exel, E.; Hoozemans, J.J.M.; Veerhuis, R.; Rozemuller, A.J.M.; van Gool, W.A. Neuroinflammation—An Early Event in Both the History and Pathogenesis of Alzheimer’s Disease. Neurodegener. Dis. 2010, 7, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, I.; Galea, I.; Perry, V.H. What Is the Blood–Brain Barrier (Not)? Trends Immunol. 2007, 28, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Palmer, T.D. Cellular Repair of CNS Disorders: An Immunological Perspective. Hum. Mol. Genet. 2008, 17, R84–R92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, A. Transplantation in the Central Nervous System. Sci. Am. 1986, 255, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.; Fujinami, R. Inflammation, Demyelination, Neurodegeneration and Neuroprotection in the Pathogenesis of Multiple Sclerosis. J. Neuroimmunol. 2007, 184, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Galea, I.; Bernardes-Silva, M.; Forse, P.A.; van Rooijen, N.; Liblau, R.S.; Perry, V.H. An Antigen-Specific Pathway for CD8 T Cells across the Blood-Brain Barrier. J. Exp. Med. 2007, 204, 2023–2030. [Google Scholar] [CrossRef] [Green Version]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted Overexpression of Human α-Synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic Inflammation in Ageing, Cardiovascular Disease, and Frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS Immune Privilege: Hiding in Plain Sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. The Ins and Outs of T-Lymphocyte Trafficking to the CNS: Anatomical Sites and Molecular Mechanisms. Trends Immunol. 2005, 26, 485–495. [Google Scholar] [CrossRef] [PubMed]
- González-Scarano, F.; Martín-García, J. The Neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.M.; Olson, K.E.; Estes, K.A.; Flanagan, K.; Gendelman, H.E.; Mosley, R.L. Dual Destructive and Protective Roles of Adaptive Immunity in Neurodegenerative Disorders. Transl. Neurodegener. 2014, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrithers, M.D.; Visintin, I.; Kang, S.J.; Janeway, C.A. Differential Adhesion Molecule Requirements for Immune Surveillance and Inflammatory Recruitment. Brain 2000, 123, 1092–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Mosley, R.L.; Reynolds, A.D.; Dhar, A.; Jackson-Lewis, V.; Gordon, P.H.; Przedborski, S.; Gendelman, H.E. Adaptive Immune Neuroprotection in G93A-SOD1 Amyotrophic Lateral Sclerosis Mice. PLoS ONE 2008, 3, e2740. [Google Scholar] [CrossRef] [Green Version]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated α–Synuclein Immunity Accelerates Degeneration of Nigral Dopaminergic Neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef] [Green Version]
- Appel, S.H. CD4+ T Cells Mediate Cytotoxicity in Neurodegenerative Diseases. J. Clin. Investig. 2009, 119, 13–15. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Investig. 2008, 92, 119–182. [Google Scholar] [CrossRef]
- Liu, J.; Gong, N.; Huang, X.; Reynolds, A.D.; Mosley, R.L.; Gendelman, H.E. Neuromodulatory Activities of CD4+ CD25+ Regulatory T Cells in a Murine Model of HIV-1-Associated Neurodegeneration. J. Immunol. 2009, 182, 3855–3865. [Google Scholar] [CrossRef] [Green Version]
- Planas, A.M.; Chamorro, A. Regulatory T Cells Protect the Brain after Stroke. Nat. Med. 2009, 15, 138–139. [Google Scholar] [CrossRef]
- Iliff, J.J.; Lee, H.; Yu, M.; Feng, T.; Logan, J.; Nedergaard, M.; Benveniste, H. Brain-Wide Pathway for Waste Clearance Captured by Contrast-Enhanced MRI. J. Clin. Investig. 2013, 123, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.; Carare, R.O.; Bechmann, I.; Flügel, A.; Laman, J.D.; Weller, R.O. Vascular, Glial, and Lymphatic Immune Gateways of the Central Nervous System. Acta Neuropathol. 2016, 132, 317–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the Functions and Relationships of the Glymphatic System and Meningeal Lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [Green Version]
- Van Zwam, M.; Huizinga, R.; Heijmans, N.; van Meurs, M.; Wierenga-Wolf, A.F.; Melief, M.-J.; Hintzen, R.Q.; ’t Hart, B.A.; Amor, S.; Boven, L.A.; et al. Surgical Excision of CNS-Draining Lymph Nodes Reduces Relapse Severity in Chronic-Relapsing Experimental Autoimmune Encephalomyelitis. J. Pathol. 2009, 217, 543–551. [Google Scholar] [CrossRef]
- Cohen, J.; Torres, C. Astrocyte Senescence: Evidence and Significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [Green Version]
- Stojiljkovic, M.R.; Ain, Q.; Bondeva, T.; Heller, R.; Schmeer, C.; Witte, O.W. Phenotypic and Functional Differences between Senescent and Aged Murine Microglia. Neurobiol. Aging 2019, 74, 56–69. [Google Scholar] [CrossRef]
- Harry, G.J. Microglia during Development and Aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia Actively Regulate the Number of Functional Synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting Microglial and Peripheral Immune Cell Crosstalk to Treat Alzheimer’s Disease. J. Neuroinflamm. 2019, 16, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s Disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic Microglia in the Aging Human Brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the Immune Response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Schwartz, M. Targeting Neuro–Immune Communication in Neurodegeneration: Challenges and Opportunities. J. Exp. Med. 2018, 215, 2702–2704. [Google Scholar] [CrossRef] [Green Version]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef]
- Yao, P.; Lin, P.; Gokoolparsadh, A.; Assareh, A.; Thang, M.W.C.; Voineagu, I. Coexpression Networks Identify Brain Region–Specific Enhancer RNAs in the Human Brain. Nat. Neurosci. 2015, 18, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, P.E.; Tan, S.; Wills, T.E.; Porritt, M.J.; Howells, D.W. Comparison of Inflammation in the Brain and Spinal Cord Following Mechanical Injury. J. Neurotrauma 2008, 25, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Wyss-Coray, T. Microglial Dysfunction in Brain Aging and Alzheimer’s Disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and Anti-Inflammaging: A Systemic Perspective on Aging and Longevity Emerged from Studies in Humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; de Los Santos, M.B.; et al. Clonally Expanded CD8 T Cells Patrol the Cerebrospinal Fluid in Alzheimer’s Disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Edwards, F.A. A Unifying Hypothesis for Alzheimer’s Disease: From Plaques to Neurodegeneration. Trends Neurosci. 2019, 42, 310–322. [Google Scholar] [CrossRef]
- Perry, V.H.; Holmes, C. Microglial Priming in Neurodegenerative Disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; el Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Wyatt-Johnson, S.K.; Brutkiewicz, R.R. The Complexity of Microglial Interactions with Innate and Adaptive Immune Cells in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 592359. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia Contributes to Plaque Growth by Cell Death Due to Uptake of Amyloid β in the Brain of Alzheimer’s Disease Mouse Model. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef]
- Friker, L.L.; Scheiblich, H.; Hochheiser, I.V.; Brinkschulte, R.; Riedel, D.; Latz, E.; Geyer, M.; Heneka, M.T. β-Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep. 2020, 30, 3743–3754.e6. [Google Scholar] [CrossRef] [PubMed]
- Frackowiak, J.; Wisniewski, H.M.; Wegiel, J.; Merz, G.S.; Iqbal, K.; Wang, K.C. Ultrastructure of the Microglia That Phagocytose Amyloid and the Microglia That Produce β-Amyloid Fibrils. Acta Neuropathol. 1992, 84, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Jantaratnotai, N.; Ryu, J.K.; Kim, S.U.; McLarnon, J.G. Amyloid β Peptide-Induced Corpus Callosum Damage and Glial Activation in Vivo. Neuroreport 2003, 14, 1429–1433. [Google Scholar] [CrossRef]
- Desai, M.K.; Guercio, B.J.; Narrow, W.C.; Bowers, W.J. An Alzheimer’s Disease-Relevant Presenilin-1 Mutation Augments Amyloid-Beta-Induced Oligodendrocyte Dysfunction. Glia 2011, 59, 627–640. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Allsop, D. Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.-E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S.B.; Grøntvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial Activation and Inflammation along the Alzheimer’s Disease Continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, S.M. Thymus Lineage Commitment: A Single Switch. Immunity 2008, 28, 297–299. [Google Scholar] [CrossRef]
- Dustin, M.L. The Cellular Context of T Cell Signaling. Immunity 2009, 30, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, F.A.; Oettgen, H.C. Adaptive Immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lee, C.H.; Qi, C.; Tailor, P.; Feng, J.; Abbasi, S.; Atsumi, T.; Morse, H.C. IRF8 Regulates B-Cell Lineage Specification, Commitment, and Differentiation. Blood 2008, 112, 4028–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding Roles for CD4+ T Cells in Immunity to Viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Byram, S.C. CD4-Positive T Cell-Mediated Neuroprotection Requires Dual Compartment Antigen Presentation. J. Neurosci. 2004, 24, 4333–4339. [Google Scholar] [CrossRef] [Green Version]
- Clausen, F.; Lorant, T.; Lewén, A.; Hillered, L. T Lymphocyte Trafficking: A Novel Target for Neuroprotection in Traumatic Brain Injury. J. Neurotrauma 2007, 24, 1295–1307. [Google Scholar] [CrossRef]
- Mundt, S.; Greter, M.; Flügel, A.; Becher, B. The CNS Immune Landscape from the Viewpoint of a T Cell. Trends Neurosci. 2019, 42, 667–679. [Google Scholar] [CrossRef]
- Dá Mesquita, S.; Ferreira, A.C.; Sousa, J.C.; Correia-Neves, M.; Sousa, N.; Marques, F. Insights on the Pathophysiology of Alzheimer’s Disease: The Crosstalk between Amyloid Pathology, Neuroinflammation and the Peripheral Immune System. Neurosci. Biobehav. Rev. 2016, 68, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Mietelska-Porowska, A.; Wojda, U. T Lymphocytes and Inflammatory Mediators in the Interplay between Brain and Blood in Alzheimer’s Disease: Potential Pools of New Biomarkers. J. Immunol. Res. 2017, 2017, 4626540. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; el Khoury, J. Microglia in Neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The Movers and Shapers in Immune Privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Malissen, B. Innate and Adaptive Immunity: Specificities and Signaling Hierarchies Revisited. Nat. Immunol. 2005, 6, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An Effector CD4 T Cell Lineage with Regulatory T Cell Ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T Cell Subsets and Their Signature Cytokines in Autoimmune and Inflammatory Diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Benveniste, E.N. Immune Function of Astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef]
- Xie, L.; Yang, S.-H. Interaction of Astrocytes and T Cells in Physiological and Pathological Conditions. Brain Res. 2015, 1623, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 Lymphocytes Promote Blood-Brain Barrier Disruption and Central Nervous System Inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [Green Version]
- Mount, M.P.; Lira, A.; Grimes, D.; Smith, P.D.; Faucher, S.; Slack, R.; Anisman, H.; Hayley, S.; Park, D.S. Involvement of Interferon-γ in Microglial-Mediated Loss of Dopaminergic Neurons. J. Neurosci. 2007, 27, 3328–3337. [Google Scholar] [CrossRef]
- Duffy, S.S.; Keating, B.A.; Perera, C.J.; Moalem-Taylor, G. The Role of Regulatory T Cells in Nervous System Pathologies. J. Neurosci. Res. 2018, 96, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Lowther, D.E.; Hafler, D.A. Regulatory T Cells in the Central Nervous System. Immunol. Rev. 2012, 248, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Deliyanti, D.; Talia, D.M.; Zhu, T.; Maxwell, M.J.; Agrotis, A.; Jerome, J.R.; Hargreaves, E.M.; Gerondakis, S.; Hibbs, M.L.; Mackay, F.; et al. Foxp3+ Tregs Are Recruited to the Retina to Repair Pathological Angiogenesis. Nat. Commun. 2017, 8, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfo, M.T.; Jang, H.R.; Bagnasco, S.M.; Ko, G.-J.; Agreda, P.; Satpute, S.R.; Crow, M.T.; King, L.S.; Rabb, H. Foxp3+ Regulatory T Cells Participate in Repair of Ischemic Acute Kidney Injury. Kidney Int. 2009, 76, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haertel, E.; Joshi, N.; Hiebert, P.; Kopf, M.; Werner, S. Regulatory T Cells Are Required for Normal and Activin-promoted Wound Repair in Mice. Eur. J. Immunol. 2018, 48, 1001–1013. [Google Scholar] [CrossRef] [Green Version]
- Rossi, B.; Angiari, S.; Zenaro, E.; Budui, S.L.; Constantin, G. Vascular Inflammation in Central Nervous System Diseases: Adhesion Receptors Controlling Leukocyte-Endothelial Interactions. J. Leukoc. Biol. 2011, 89, 539–556. [Google Scholar] [CrossRef]
- Kivisäkk, P.; Tucky, B.; Wei, T.; Campbell, J.J.; Ransohoff, R.M. Human Cerebrospinal Fluid Contains CD4+ Memory T Cells Expressing Gut- or Skin-Specific Trafficking Determinants: Relevance for Immunotherapy. BMC Immunol. 2006, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-Dimensional, Single-Cell Characterization of the Brain’s Immune Compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef]
- Machhi, J.; Kevadiya, B.D.; Muhammad, I.K.; Herskovitz, J.; Olson, K.E.; Mosley, R.L.; Gendelman, H.E. Harnessing Regulatory T Cell Neuroprotective Activities for Treatment of Neurodegenerative Disorders. Mol. Neurodegener. 2020, 15, 32. [Google Scholar] [CrossRef]
- Amor, S.; Woodroofe, M.N. Innate and Adaptive Immune Responses in Neurodegeneration and Repair. Immunology 2014, 141, 287–291. [Google Scholar] [CrossRef]
- Goverman, J. Autoimmune T Cell Responses in the Central Nervous System. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, N.; Lassmann, S.; Li, Z.; Odoardi, F.; Ritter, T.; Ziemssen, T.; Klinkert, W.E.F.; Ellwart, J.W.; Bradl, M.; Krivacic, K.; et al. The Activation Status of Neuroantigen-Specific T Cells in the Target Organ Determines the Clinical Outcome of Autoimmune Encephalomyelitis. J. Exp. Med. 2004, 199, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Bartholomäus, I.; Kawakami, N.; Odoardi, F.; Schläger, C.; Miljkovic, D.; Ellwart, J.W.; Klinkert, W.E.F.; Flügel-Koch, C.; Issekutz, T.B.; Wekerle, H.; et al. Effector T Cell Interactions with Meningeal Vascular Structures in Nascent Autoimmune CNS Lesions. Nature 2009, 462, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zheng, H. Peripheral Immune System in Aging and Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 51. [Google Scholar] [CrossRef]
- Cacabelos, R.; Fernandez-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular Genetics of Alzheimer’s Disease and Aging. Methods Find. Exp. Clin. Pharm. 2005, 27 (Suppl. SA), 1–573. [Google Scholar]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 599. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.C.; Cai, H.; Borchelt, D.R.; Price, D.L. Genetically Engineered Mouse Models of Neurodegenerative Diseases. Nat. Neurosci. 2002, 5, 633–639. [Google Scholar] [CrossRef]
- Bloom, F.E.; Reilly, J.F.; Redwine, J.M.; Wu, C.-C.; Young, W.G.; Morrison, J.H. Mouse Models of Human Neurodegenerative Disorders. Arch. Neurol. 2005, 62, 185. [Google Scholar] [CrossRef]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal Models of Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Janus, C.; Welzl, H. Mouse Models of Neurodegenerative Diseases: Criteria and General Methodology. In Mouse Models for Drug Discovery; Humana Press: Totowa, NJ, USA, 2010; pp. 323–345. [Google Scholar]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T Cells from Patients with Parkinson’s Disease Recognize α-Synuclein Peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Lodygin, D.; Hermann, M.; Schweingruber, N.; Flügel-Koch, C.; Watanabe, T.; Schlosser, C.; Merlini, A.; Körner, H.; Chang, H.-F.; Fischer, H.J.; et al. β-Synuclein-Reactive T Cells Induce Autoimmune CNS Grey Matter Degeneration. Nature 2019, 566, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.-A.; Mills, K.H.G.; Lynch, M.A. IFN-γ Production by Amyloid β–Specific Th1 Cells Promotes Microglial Activation and Increases Plaque Burden in a Mouse Model of Alzheimer’s Disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Town, T.; Tan, J.; Flavell, R.A.; Mullan, M. T-Cells in Alzheimer’s Disease. Neuromol. Med. 2005, 7, 255–264. [Google Scholar] [CrossRef]
- Li, M.; Shang, D.-S.; Zhao, W.-D.; Tian, L.; Li, B.; Fang, W.-G.; Zhu, L.; Man, S.-M.; Chen, Y.-H. Amyloid β Interaction with Receptor for Advanced Glycation End Products Up-Regulates Brain Endothelial CCR5 Expression and Promotes T Cells Crossing the Blood-Brain Barrier. J. Immunol. 2009, 182, 5778–5788. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, M.T.; Merlini, M.; Späni, C.; Gericke, C.; Schweizer, N.; Enzmann, G.; Engelhardt, B.; Kulic, L.; Suter, T.; Nitsch, R.M. T-Cell Brain Infiltration and Immature Antigen-Presenting Cells in Transgenic Models of Alzheimer’s Disease-like Cerebral Amyloidosis. Brain Behav. Immun. 2016, 54, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Lueg, G.; Gross, C.C.; Lohmann, H.; Johnen, A.; Kemmling, A.; Deppe, M.; Groger, J.; Minnerup, J.; Wiendl, H.; Meuth, S.G.; et al. Clinical Relevance of Specific T-Cell Activation in the Blood and Cerebrospinal Fluid of Patients with Mild Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 81–89. [Google Scholar] [CrossRef]
- Rogers, J.; Luber-Narod, J.; Styren, S.D.; Civin, W.H. Expression of Immune System-Associated Antigens by Cells of the Human Central Nervous System: Relationship to the Pathology of Alzheimer’s Disease. Neurobiol. Aging 1988, 9, 339–349. [Google Scholar] [CrossRef]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T Cells in the Brain of Alzheimer’s Disease and Other Neurological Diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Itagaki, S.; McGeer, P.L.; Akiyama, H. Presence of T-Cytotoxic Suppressor and Leucocyte Common Antigen Positive Cells in Alzheimer’s Disease Brain Tissue. Neurosci. Lett. 1988, 91, 259–264. [Google Scholar] [CrossRef]
- Pirttilä, T.; Mattinen, S.; Frey, H. The Decrease of CD8-Positive Lymphocytes in Alzheimer’s Disease. J. Neurol. Sci. 1992, 107, 160–165. [Google Scholar] [CrossRef]
- Merlini, M.; Kirabali, T.; Kulic, L.; Nitsch, R.M.; Ferretti, M.T. Extravascular CD3+ T Cells in Brains of Alzheimer Disease Patients Correlate with Tau but Not with Amyloid Pathology: An Immunohistochemical Study. Neurodegener. Dis. 2018, 18, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Parachikova, A.; Agadjanyan, M.G.; Cribbs, D.H.; Blurton-Jones, M.; Perreau, V.; Rogers, J.; Beach, T.G.; Cotman, C.W. Inflammatory Changes Parallel the Early Stages of Alzheimer Disease. Neurobiol. Aging 2007, 28, 1821–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, J.A.H.; Estes, K.A.; Kosloski, L.M.; Allen, H.E.; Dempsey, K.M.; Torres-Russotto, D.R.; Meza, J.L.; Santamaria, P.M.; Bertoni, J.M.; Murman, D.L.; et al. CD4+ Regulatory and Effector/Memory T Cell Subsets Profile Motor Dysfunction in Parkinson’s Disease. J. Neuroimmune Pharmacol. 2012, 7, 927–938. [Google Scholar] [CrossRef] [Green Version]
- Gendelman, H.E.; Mosley, R.L. A Perspective on Roles Played by Innate and Adaptive Immunity in the Pathobiology of Neurodegenerative Disorders. J. Neuroimmune Pharmacol. 2015, 10, 645–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s Disease Patients Have a Complex Phenotypic and Functional Th1 Bias: Cross-Sectional Studies of CD4+ Th1/Th2/T17 and Treg in Drug-Naïve and Drug-Treated Patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Shalit, F.; Sredni, B.; Brodie, C.; Kott, E.; Huberman, M. T Lymphocyte Subpopulations and Activation Markers Correlate with Severity of Alzheimer’s Disease. Clin. Immunol. Immunopathol. 1995, 75, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Calabrese, E.; Marventano, I.; Piancone, F.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. Increased Activity of Th-17 and Th-9 Lymphocytes and a Skewing of the Post-Thymic Differentiation Pathway Are Seen in Alzheimer’s Disease. Brain Behav. Immun. 2011, 25, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Agnes, P.; Christiane, S.; Peter, D. T-Cells Show Increased Production of Cytokines and Activation Markers in Alzheimer’s Disease. Brain. Disord. Ther. 2013, 3, 3–112. [Google Scholar]
- Ciccocioppo, F.; Lanuti, P.; Pierdomenico, L.; Simeone, P.; Bologna, G.; Ercolino, E.; Buttari, F.; Fantozzi, R.; Thomas, A.; Onofrj, M.; et al. The Characterization of Regulatory T-Cell Profiles in Alzheimer’s Disease and Multiple Sclerosis. Sci. Rep. 2019, 9, 8788. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; O’Banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory Processes in Alzheimer’s Disease. J. Neural Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef]
- Zhang, J.; Ke, K.-F.; Liu, Z.; Qiu, Y.-H.; Peng, Y.-P. Th17 Cell-Mediated Neuroinflammation Is Involved in Neurodegeneration of Aβ1-42-Induced Alzheimer’s Disease Model Rats. PLoS ONE 2013, 8, e75786. [Google Scholar] [CrossRef] [PubMed]
- Chui, R.; Dorovini-Zis, K. Regulation of CCL2 and CCL3 Expression in Human Brain Endothelial Cells by Cytokines and Lipopolysaccharide. J. Neuroinflamm. 2010, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, T.J.; Taha, L.; Spitzer, P.; Hellstern, J.; Herrmann, M.; Kornhuber, J.; Maler, J.M. Imbalance of Circulating Th17 and Regulatory T Cells in Alzheimer’s Disease: A Case Control Study. Front. Immunol. 2018, 9, 1213. [Google Scholar] [CrossRef] [Green Version]
- Baek, H.; Ye, M.; Kang, G.-H.; Lee, C.; Lee, G.; Choi, D.B.; Jung, J.; Kim, H.; Lee, S.; Kim, J.S.; et al. Neuroprotective Effects of CD4+CD25+Foxp3+ Regulatory T Cells in a 3xTg-AD Alzheimer’s Disease Model. Oncotarget 2016, 7, 69347–69357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dansokho, C.; Ait Ahmed, D.; Aid, S.; Toly-Ndour, C.; Chaigneau, T.; Calle, V.; Cagnard, N.; Holzenberger, M.; Piaggio, E.; Aucouturier, P.; et al. Regulatory T Cells Delay Disease Progression in Alzheimer-like Pathology. Brain 2016, 139, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.C.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ Regulatory T Cells Induce Alternative Activation of Human Monocytes/Macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, H.; Xie, Z.; Wei, L.; Bi, J. Systemic Transplantation of Human Umbilical Cord Derived Mesenchymal Stem Cells-Educated T Regulatory Cells Improved the Impaired Cognition in AβPPswe/PS1dE9 Transgenic Mice. PLoS ONE 2013, 8, e69129. [Google Scholar] [CrossRef] [Green Version]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou-Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking Immune Tolerance by Targeting Foxp3+ Regulatory T Cells Mitigates Alzheimer’s Disease Pathology. Nat. Commun. 2015, 6, 7967. [Google Scholar] [CrossRef] [Green Version]
- Mayne, K.; White, J.A.; McMurran, C.E.; Rivera, F.J.; de la Fuente, A.G. Aging and Neurodegenerative Disease: Is the Adaptive Immune System a Friend or Foe? Front. Aging Neurosci. 2020, 12, 572090. [Google Scholar] [CrossRef]
- Marsh, S.E.; Abud, E.M.; Lakatos, A.; Karimzadeh, A.; Yeung, S.T.; Davtyan, H.; Fote, G.M.; Lau, L.; Weinger, J.G.; Lane, T.E.; et al. The Adaptive Immune System Restrains Alzheimer’s Disease Pathogenesis by Modulating Microglial Function. Proc. Natl. Acad. Sci. USA 2016, 113, E1316–E1325. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Princiotta Cariddi, L.; Mauri, M.; Cosentino, M.; Versino, M.; Marino, F. Alzheimer’s Disease: From Immune Homeostasis to Neuroinflammatory Condition. Int. J. Mol. Sci. 2022, 23, 13008. https://doi.org/10.3390/ijms232113008
Princiotta Cariddi L, Mauri M, Cosentino M, Versino M, Marino F. Alzheimer’s Disease: From Immune Homeostasis to Neuroinflammatory Condition. International Journal of Molecular Sciences. 2022; 23(21):13008. https://doi.org/10.3390/ijms232113008
Chicago/Turabian StylePrinciotta Cariddi, Lucia, Marco Mauri, Marco Cosentino, Maurizio Versino, and Franca Marino. 2022. "Alzheimer’s Disease: From Immune Homeostasis to Neuroinflammatory Condition" International Journal of Molecular Sciences 23, no. 21: 13008. https://doi.org/10.3390/ijms232113008