
Fatty Acid Amide Hydrolase Deficiency Is Associated with Deleterious Cardiac Effects after Myocardial Ischemia and Reperfusion in Mice

 , , ,
, , ,  and
and 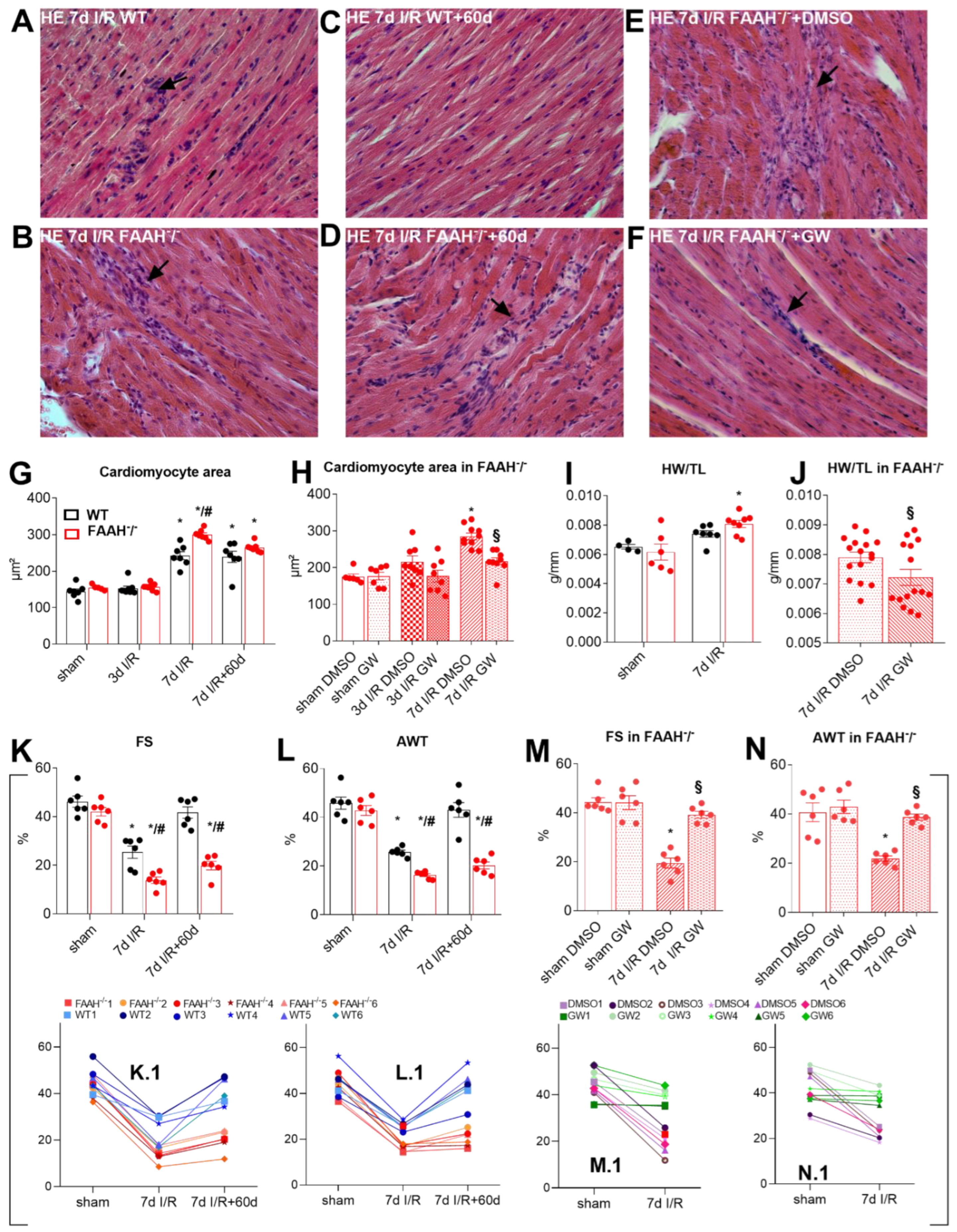{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Problem Definition
1.2. Study Motivation
1.3. Aims and Objective
2. Results
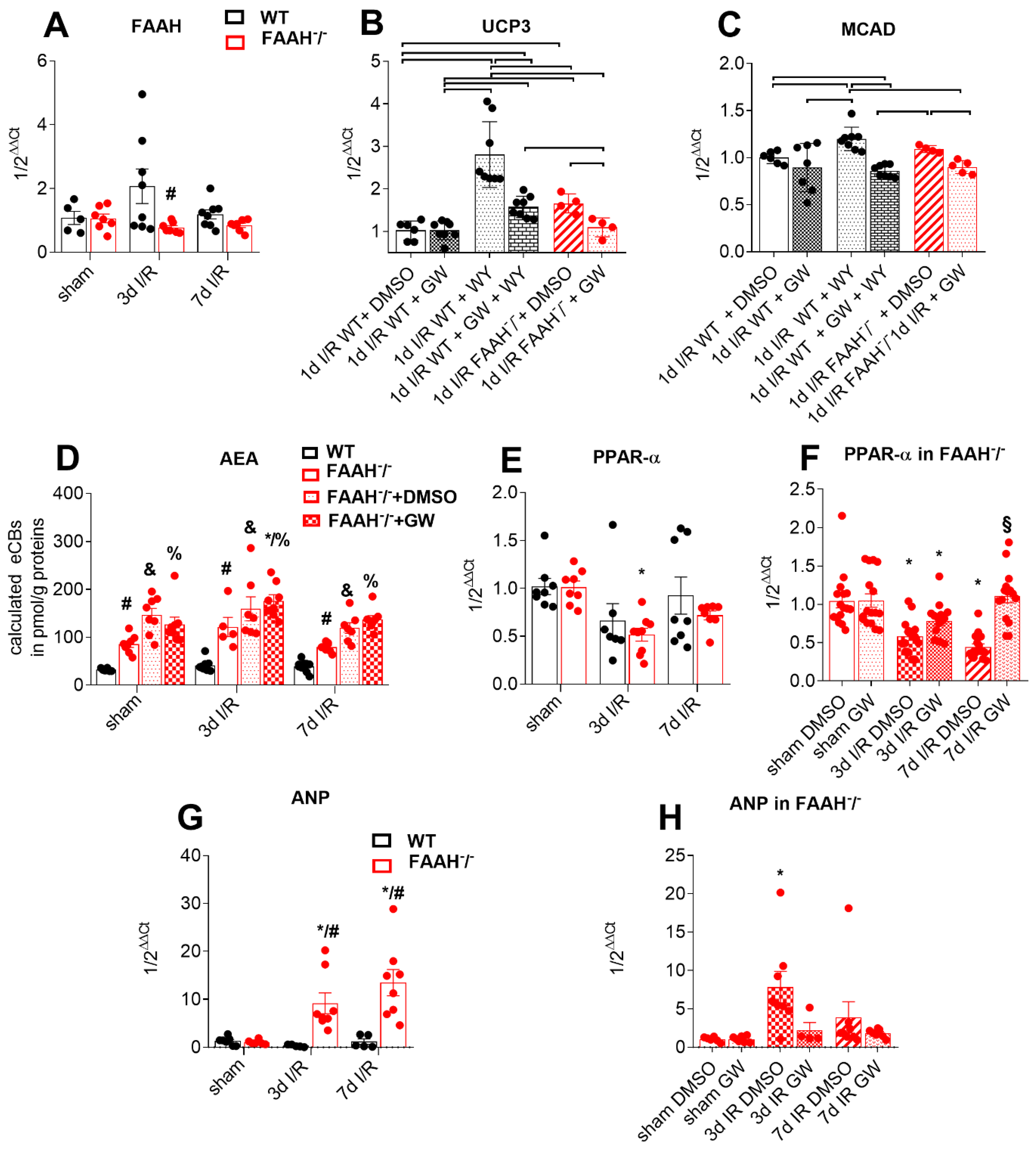
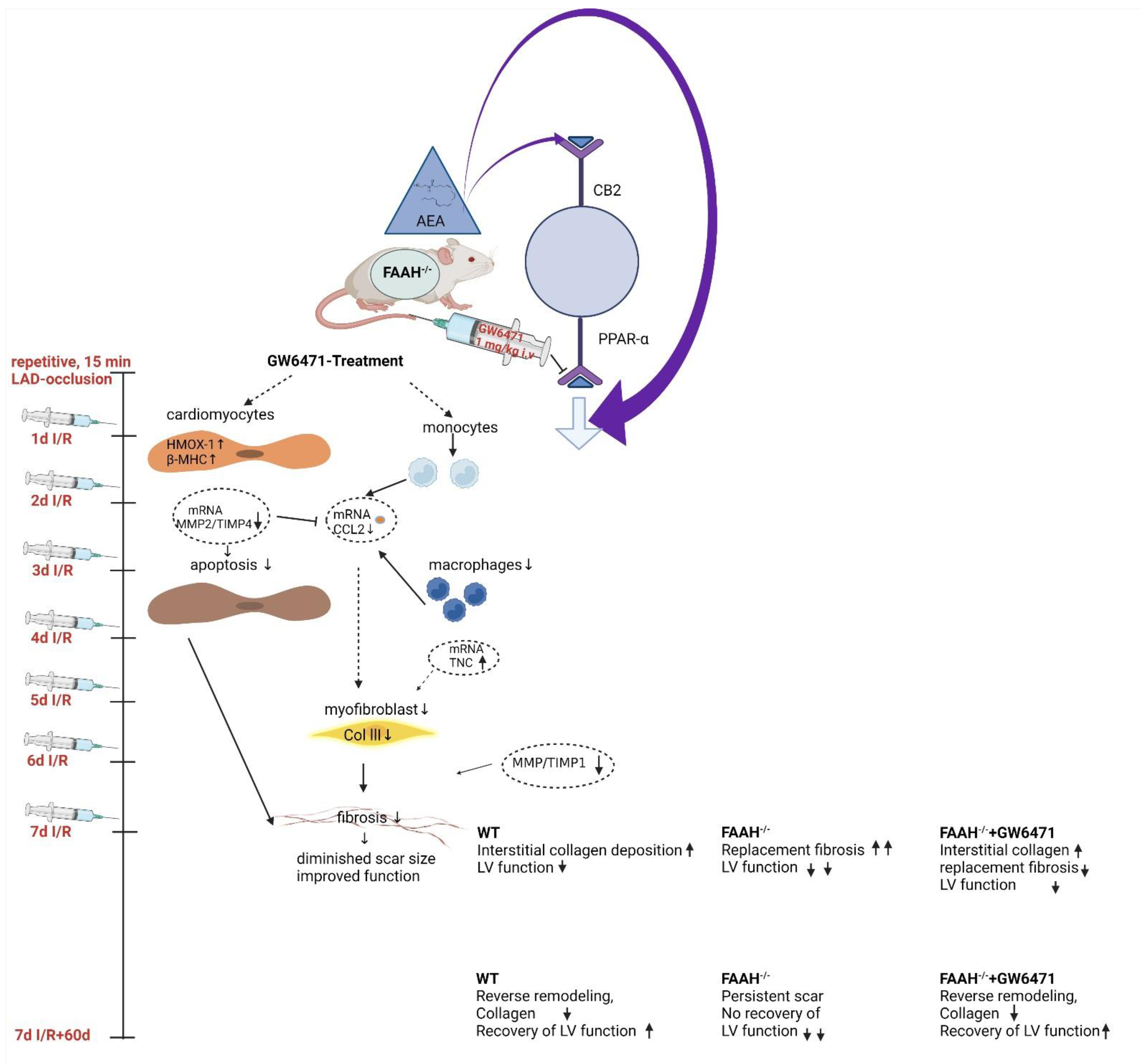
2.1. Fatty Acid Amide Hydrolase Deficient Mice Show Persistent Infiltration, Hypertrophy, and Loss of Function after I/R but Recover after PPAR-α Inhibition with GW6471
2.2. Proof of Principle
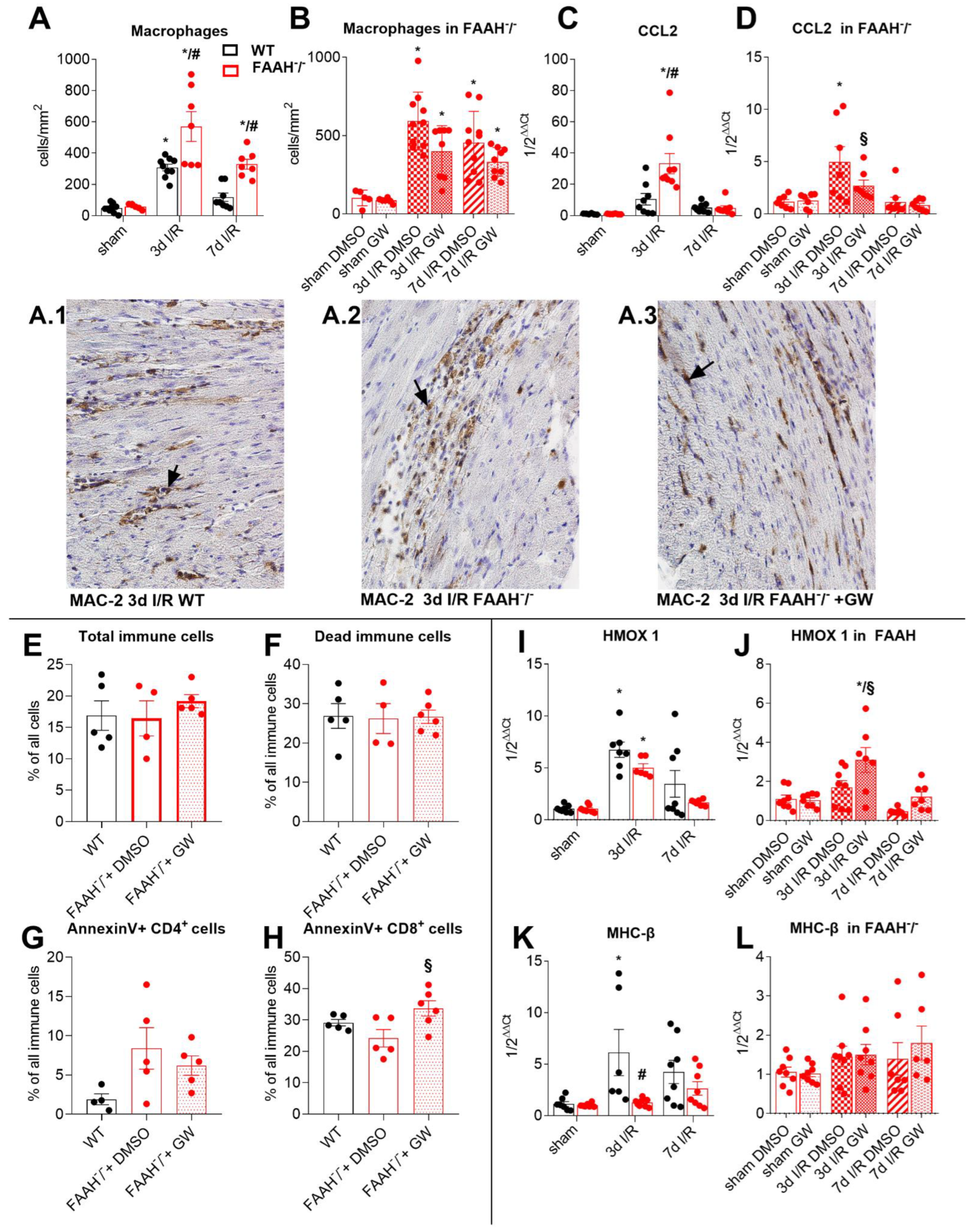
2.3. Fatty Acid Amide Hydrolase Deficiency Modulates Inflammation and Adaptation to Ischemia and Reperfusion
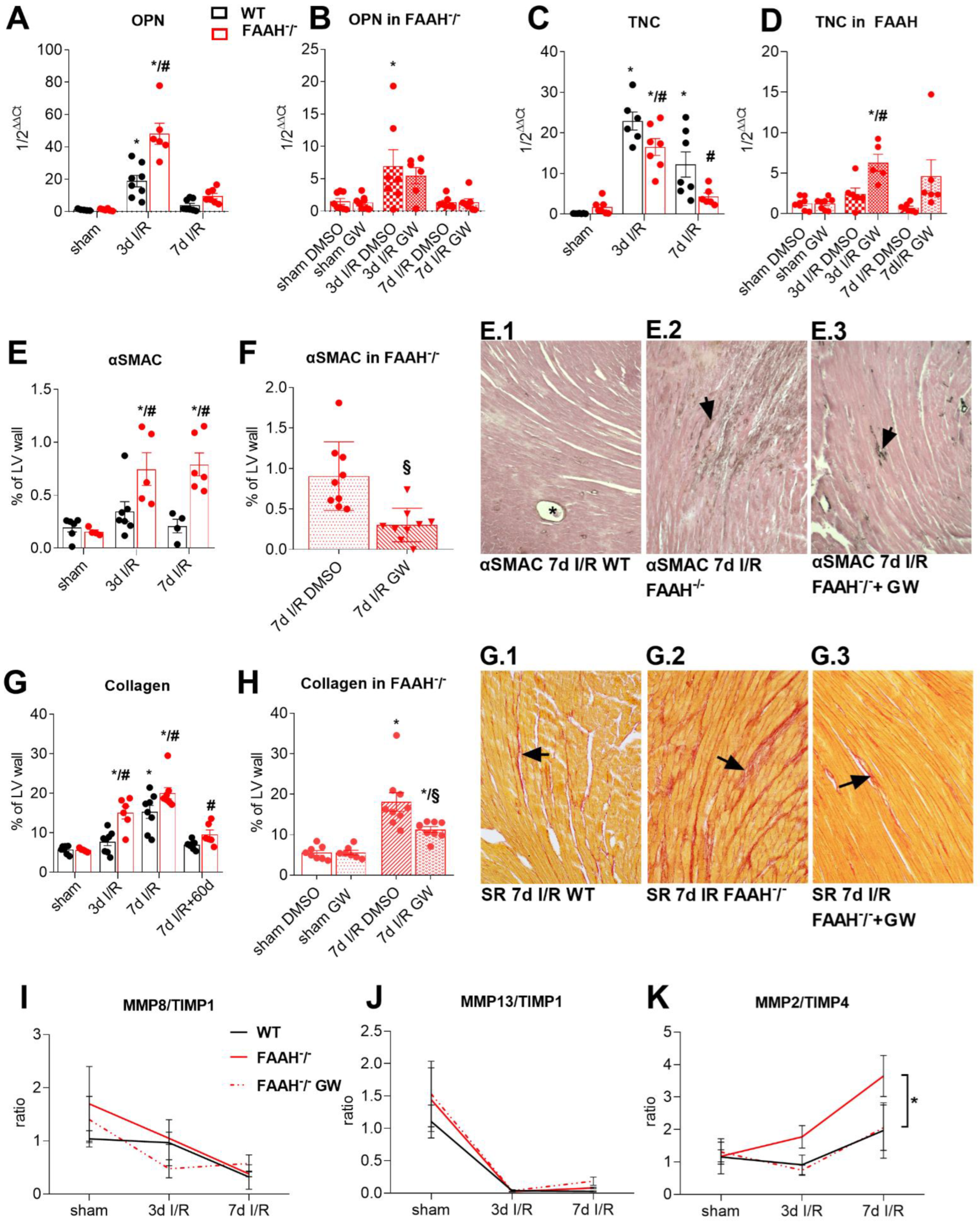
2.4. Fatty Acid Amide Hydrolase Deficiency Induces Hypertrophy and Increases Remodeling with High Myofibroblasts Accumulation and Collagen Deposition
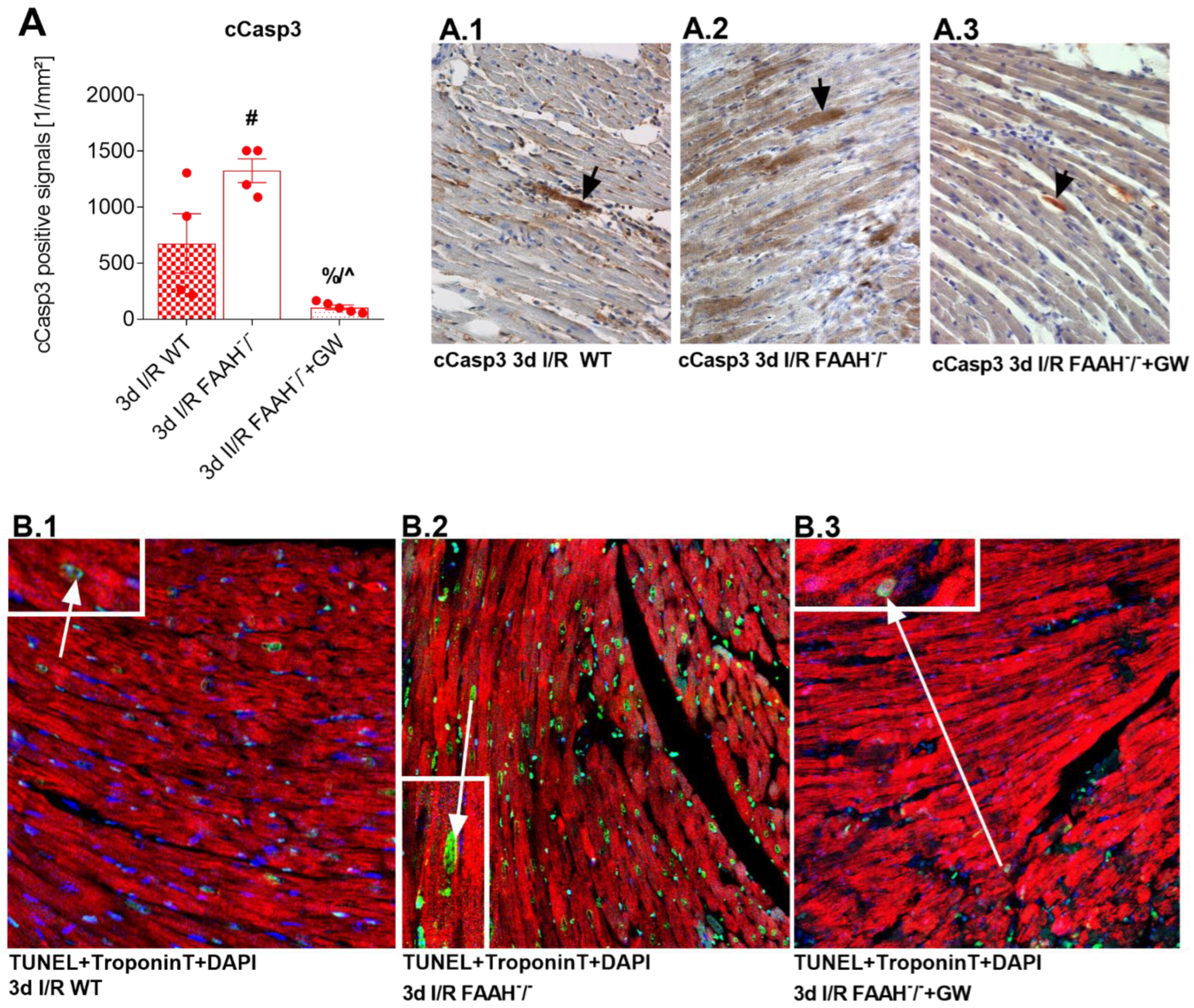
2.5. PPAR-α Antagonist Reduces Enhanced Apoptosis and Cardiomyocyte Loss in FAAH−/− Mice to WT Levels
3. Discussion
Limitation of the Study
4. Materials and Methods
4.1. Study Animals
4.2. Brief Repetitive I/R Protocol
4.3. Assessment of Left Ventricular Function—M-Mode Echocardiography
4.4. Treatment with PPAR-α Agonist WY14.634 in WT and PPAR-α Agonism with GW6471
4.5. Hypertrophy
4.6. Histology
4.7. Immunohistochemistry and Immunofluorescence
4.8. Triple-Immunofluorescence Staining for Cardiomyocytes(Troponin T), TUNEL, and DAPI
4.9. Flow Cytometry Analysis
4.10. Endocannabinoid Measurements by LC-MS/MS
4.11. Gene Expression Analysis
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. American Heart Association Council on, E.; Prevention Statistics, C.; Stroke Statistics, S. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 SDG Collaborators. Department of Error. Lancet 2017, 389, e1. [Google Scholar] [CrossRef] [Green Version]
- Kalra, D.K.; Zoghbi, W.A. Myocardial hibernation in coronary artery disease. Curr. Atheroscler. Rep. 2002, 4, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, Y.; Cavanaugh, S.M.; Dhamoon, A.S. Myocardial stunning and hibernation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Heusch, G.; Schulz, R.; Rahimtoola, S.H. Myocardial hibernation: A delicate balance. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H984–H999. [Google Scholar] [CrossRef] [Green Version]
- Dewald, O.; Frangogiannis, N.G.; Zoerlein, M.; Duerr, G.D.; Klemm, C.; Knuefermann, P.; Taffet, G.; Michael, L.H.; Crapo, J.D.; Welz, A.; et al. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc. Natl. Acad. Sci. USA 2003, 100, 2700–2705. [Google Scholar] [CrossRef] [Green Version]
- Duerr, G.D.; Heinemann, J.C.; Suchan, G.; Kolobara, E.; Wenzel, D.; Geisen, C.; Matthey, M.; Passe-Tietjen, K.; Mahmud, W.; Ghanem, A.; et al. The endocannabinoid-CB2 receptor axis protects the ischemic heart at the early stage of cardiomyopathy. Basic Res. Cardiol. 2014, 109, 425. [Google Scholar] [CrossRef]
- Duerr, G.D.; Heinemann, J.C.; Gestrich, C.; Heuft, T.; Klaas, T.; Keppel, K.; Roell, W.; Klein, A.; Zimmer, A.; Velten, M.; et al. Impaired border zone formation and adverse remodeling after reperfused myocardial infarction in cannabinoid CB2 receptor deficient mice. Life Sci. 2015, 138, 8–17. [Google Scholar] [CrossRef]
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; Piscitelli, F. The Endocannabinoid System and its Modulation by Phytocannabinoids. Neurotherapeutics 2015, 12, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Di Filippo, C.; Rossi, F.; Rossi, S.; D’Amico, M. Cannabinoid CB2 receptor activation reduces mouse myocardial ischemia-reperfusion injury: Involvement of cytokine/chemokines and PMN. J. Leukoc. Biol. 2004, 75, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Jiang, L.S.; Bu, J.; Huang, X.J.; Song, W.; Du, Y.P.; He, B. Cannabinoid-2 receptor activation protects against infarct and ischemia-reperfusion heart injury. J. Cardiovasc. Pharmacol. 2012, 59, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, F.; Zhang, Y.M.; Zhou, J.J.; Zhang, Y. Activation of cannabinoid type 2 receptor by JWH133 protects heart against ischemia/reperfusion-induced apoptosis. Cell. Physiol. Biochem. 2013, 31, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The pharmacology of cannabinoid receptors and their ligands: An overview. Int. J. Obes. 2006, 30 (Suppl. 1), S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Lamontagne, D.; Lepicier, P.; Lagneux, C.; Bouchard, J.F. The endogenous cardiac cannabinoid system: A new protective mechanism against myocardial ischemia. Arch. Mal. Coeur Vaiss. 2006, 99, 242–246. [Google Scholar]
- Batkai, S.; Rajesh, M.; Mukhopadhyay, P.; Hasko, G.; Liaudet, L.; Cravatt, B.F.; Csiszar, A.; Ungvari, Z.; Pacher, P. Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression, and apoptosis in mice lacking fatty acid amide hydrolase. Am. J. Physiology. Heart Circ. Physiol. 2007, 293, H909–H918. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Horvath, B.; Rajesh, M.; Matsumoto, S.; Saito, K.; Batkai, S.; Patel, V.; Tanchian, G.; Gao, R.Y.; Cravatt, B.F.; et al. Fatty acid amide hydrolase is a key regulator of endocannabinoid-induced myocardial tissue injury. Free Radic. Biol. Med. 2011, 50, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Cravatt, B.F. Characterization and manipulation of the acyl chain selectivity of fatty acid amide hydrolase. Biochemistry 2001, 40, 6107–6115. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Bennett, A. Cannabinoids: A new group of agonists of PPARs. PPAR Res. 2007, 2007, 23513. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewald, O.; Sharma, S.; Adrogue, J.; Salazar, R.; Duerr, G.D.; Crapo, J.D.; Entman, M.L.; Taegtmeyer, H. Downregulation of peroxisome proliferator-activated receptor-alpha gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation 2005, 112, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Duerr, G.D.; Heinemann, J.C.; Arnoldi, V.; Feisst, A.; Kley, J.; Ghanem, A.; Welz, A.; Dewald, O. Cardiomyocyte specific peroxisome proliferator-activated receptor-alpha overexpression leads to irreversible damage in ischemic murine heart. Life Sci. 2014, 102, 88–97. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [Green Version]
- Dyck, J.R.; Hopkins, T.A.; Bonnet, S.; Michelakis, E.D.; Young, M.E.; Watanabe, M.; Kawase, Y.; Jishage, K.; Lopaschuk, G.D. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 2006, 114, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Invest. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Barger, P.M.; Kelly, D.P. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc. Med. 2000, 10, 238–245. [Google Scholar] [CrossRef]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Invest. 2006, 116, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xiao, G.; Trujillo, C.; Chang, V.; Blanco, L.; Joseph, S.B.; Bassilian, S.; Saad, M.F.; Tontonoz, P.; Lee, W.N.; et al. Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J. Biol. Chem. 2002, 277, 50237–50244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Shen, W.J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-alpha. Future Cardiol. 2017, 13, 259–278. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.M.; Kozak, R.; Wagner, A.; Altarejos, J.Y.; Dyck, J.R.; Belke, D.D.; Severson, D.L.; Kelly, D.P.; Lopaschuk, G.D. A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: Reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J. Biol. Chem. 2002, 277, 4098–4103. [Google Scholar] [PubMed] [Green Version]
- Liu, F.; Song, R.; Feng, Y.; Guo, J.; Chen, Y.; Zhang, Y.; Chen, T.; Wang, Y.; Huang, Y.; Li, C.Y.; et al. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor alpha. Circulation 2015, 131, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Duerr, G.D.; Heinemann, J.C.; Kley, J.; Eichhorn, L.; Frede, S.; Weisheit, C.; Wehner, S.; Bindila, L.; Lutz, B.; Zimmer, A.; et al. Myocardial maladaptation to pressure overload in CB2 receptor-deficient mice. J. Mol. Cell. Cardiol. 2019, 133, 86–98. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147, S163–S171. [Google Scholar] [CrossRef] [Green Version]
- Wise, L.E.; Harloe, J.P.; Lichtman, A.H. Fatty acid amide hydrolase (FAAH) knockout mice exhibit enhanced acquisition of an aversive, but not of an appetitive, Barnes maze task. Neurobiol. Learn. Mem. 2009, 92, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Wang, H.; Wu, Q. Atrial natriuretic peptide in cardiovascular biology and disease (NPPA). Gene 2015, 569, 1–6. [Google Scholar] [CrossRef] [Green Version]
- O’sullivan, S.; Kendall, D. Cannabinoid activation of peroxisome proliferator-activated receptors: Potential for modulation of inflammatory disease. Immunobiology 2010, 215, 611–616. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hu, C.; Du, Y.; Zhang, J.; Liu, J.; Han, H.; Zhao, Y. Significant association between admission serum monocyte chemoattractant protein-1 and early changes in myocardial function in patients with first ST-segment elevation myocardial infarction after primary percutaneous coronary intervention. BMC Cardiovasc. Disord. 2019, 19, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lemos, J.A.; Morrow, D.A.; Sabatine, M.S.; Murphy, S.A.; Gibson, C.M.; Antman, E.M.; McCabe, C.H.; Cannon, C.P.; Braunwald, E. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation 2003, 107, 690–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clanton, T.L. Hypoxia-induced reactive oxygen species formation in skeletal muscle. J. Appl. Physiol. (1985) 2007, 102, 2379–2388. [Google Scholar] [CrossRef] [Green Version]
- Elbirt, K.K.; Bonkovsky, H.L. Heme oxygenase: Recent advances in understanding its regulation and role. Proc. Assoc. Am. Physicians 1999, 111, 438–447. [Google Scholar] [CrossRef]
- Duerr, G.D.; Wu, S.; Schneider, M.L.; Marggraf, V.; Weisheit, C.K.; Velten, M.; Verfuerth, L.; Frede, S.; Boehm, O.; Treede, H.; et al. CpG postconditioning after reperfused myocardial infarction is associated with modulated inflammation, less apoptosis, and better left ventricular function. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H995–H1007. [Google Scholar] [CrossRef]
- Pope, B.; Hoh, J.; Weeds, A. The ATPase activities of rat cardiac myosin isoenzymes. FEBS Lett. 1980, 118, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Holubarsch, C.; Goulette, R.P.; Litten, R.Z.; Martin, B.J.; Mulieri, L.A.; Alpert, N.R. The economy of isometric force development, myosin isoenzyme pattern and myofibrillar ATPase activity in normal and hypothyroid rat myocardium. Circ. Res. 1985, 56, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Razeghi, P.; Essop, M.F.; Huss, J.M.; Abbasi, S.; Manga, N.; Taegtmeyer, H. Hypoxia-induced switches of myosin heavy chain iso-gene expression in rat heart. Biochem. Biophys. Res. Commun. 2003, 303, 1024–1027. [Google Scholar] [CrossRef]
- Swynghedauw, B. Developmental and functional adaptation of contractile proteins in cardiac and skeletal muscles. Physiol. Rev. 1986, 66, 710–771. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, J.C.; Hewett, T.E.; Factor, S.M.; Vikstrom, K.L.; Robbins, J.; Leinwand, L.A. Expression of the beta (slow)-isoform of MHC in the adult mouse heart causes dominant-negative functional effects. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H412–H419. [Google Scholar] [CrossRef] [PubMed]
- Duerr, G.D.; Dewald, D.; Schmitz, E.J.; Verfuerth, L.; Keppel, K.; Peigney, C.; Ghanem, A.; Welz, A.; Dewald, O. Metallothioneins 1 and 2 Modulate Inflammation and Support Remodeling in Ischemic Cardiomyopathy in Mice. Mediat. Inflamm. 2016, 2016, 7174127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.D.; Mitchell, M.D.; Long, C.S. Pro-inflammatory cytokines and cardiac extracellular matrix: Regulation of fibroblast phenotype. In Interstitial Fibrosis in Heart Failure; Springer: Berlin/Heidelberg, Germany, 2005; pp. 57–81. [Google Scholar]
- Tyagi, S.C.; Kumar, S.G.; Banks, J.; Fortson, W. Co-expression of tissue inhibitor and matrix metalloproteinase in myocardium. J. Mol. Cell. Cardiol. 1995, 27, 2177–2189. [Google Scholar] [CrossRef]
- Rutschow, S.; Leschka, S.; Westermann, D.; Puhl, K.; Weitz, A.; Ladyszenskij, L.; Jaeger, S.; Zeichhardt, H.; Noutsias, M.; Schultheiss, H.P.; et al. Left ventricular enlargement in coxsackievirus-B3 induced chronic myocarditis--ongoing inflammation and an imbalance of the matrix degrading system. Eur. J. Pharmacol. 2010, 630, 145–151. [Google Scholar] [CrossRef]
- Peterson, J.T.; Li, H.; Dillon, L.; Bryant, J.W. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc. Res. 2000, 46, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Woessner, J.F., Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991, 5, 2145–2154. [Google Scholar] [CrossRef] [Green Version]
- Rohde, L.E.; Ducharme, A.; Arroyo, L.H.; Aikawa, M.; Sukhova, G.H.; Lopez-Anaya, A.; McClure, K.F.; Mitchell, P.G.; Libby, P.; Lee, R.T. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation 1999, 99, 3063–3070. [Google Scholar] [CrossRef] [Green Version]
- Spinale, F.G.; Coker, M.L.; Krombach, S.R.; Mukherjee, R.; Hallak, H.; Houck, W.V.; Clair, M.J.; Kribbs, S.B.; Johnson, L.L.; Peterson, J.T.; et al. Matrix metalloproteinase inhibition during the development of congestive heart failure: Effects on left ventricular dimensions and function. Circ. Res. 1999, 85, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Bourboulia, D.; Stetler-Stevenson, W.G. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol. 2010, 20, 161–168. [Google Scholar] [CrossRef]
- Wetzl, V.; Tiede, S.L.; Faerber, L.; Weissmann, N.; Schermuly, R.T.; Ghofrani, H.A.; Gall, H. Plasma MMP2/TIMP4 ratio at follow-up assessment predicts disease progression of idiopathic pulmonary arterial hypertension. Lung 2017, 195, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Orozco Morales, M.L.; Rinaldi, C.A.; de Jong, E.; Lansley, S.M.; Gummer, J.P.A.; Olasz, B.; Nambiar, S.; Hope, D.E.; Casey, T.H.; Lee, Y.C.G.; et al. PPARalpha and PPARgamma activation is associated with pleural mesothelioma invasion but therapeutic inhibition is ineffective. iScience 2022, 25, 103571. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Collino, M.; Castiglia, S.; Fantozzi, R.; Thiemermann, C. Activation of peroxisome proliferator-activated receptor-beta/delta attenuates myocardial ischemia/reperfusion injury in the rat. Shock 2010, 34, 117–124. [Google Scholar] [CrossRef] [PubMed]
- van den Broek, L.J.; van de Vijver, M.J. Assessment of problems in diagnostic and research immunohistochemistry associated with epitope instability in stored paraffin sections. Appl. Immunohistochem. Mol. Morphol. 2000, 8, 316–321. [Google Scholar]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [Green Version]
- Wayman, N.S.; Hattori, Y.; McDonald, M.C.; Mota-Filipe, H.; Cuzzocrea, S.; Pisano, B.; Chatterjee, P.K.; Thiemermann, C. Ligands of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J. 2002, 16, 1027–1040. [Google Scholar] [CrossRef] [Green Version]
- Esposito, E.; Rinaldi, B.; Mazzon, E.; Donniacuo, M.; Impellizzeri, D.; Paterniti, I.; Capuano, A.; Bramanti, P.; Cuzzocrea, S. Anti-inflammatory effect of simvastatin in an experimental model of spinal cord trauma: Involvement of PPAR-alpha. J. Neuroinflammation 2012, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, R.D.; Houng, A.K.; Gladysheva, I.P.; Fan, T.M.; Tripathi, R.; Reed, G.L.; Wang, D. Corin Overexpression Reduces Myocardial Infarct Size and Modulates Cardiomyocyte Apoptotic Cell Death. Int. J. Mol. Sci. 2020, 21, 3456. [Google Scholar] [CrossRef]
- Weisheit, C.; Zhang, Y.; Faron, A.; Kopke, O.; Weisheit, G.; Steinstrasser, A.; Frede, S.; Meyer, R.; Boehm, O.; Hoeft, A.; et al. Ly6C(low) and not Ly6C(high) macrophages accumulate first in the heart in a model of murine pressure-overload. PLoS ONE 2014, 9, e112710. [Google Scholar] [CrossRef]
- Duerr, G.D.; Elhafi, N.; Bostani, T.; Ellinger, J.; Swieny, L.; Kolobara, E.; Welz, A.; Dewald, O. Comparison of Myocardial Remodeling between Cryoinfarction and Reperfused Infarction in Mice. J. Biomed. Biotechnol. 2011, 2011, 961298. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajlic, S.; Surmann, L.; Zimmermann, P.; Weisheit, C.K.; Bindila, L.; Treede, H.; Velten, M.; Daiber, A.; Duerr, G.D. Fatty Acid Amide Hydrolase Deficiency Is Associated with Deleterious Cardiac Effects after Myocardial Ischemia and Reperfusion in Mice. Int. J. Mol. Sci. 2022, 23, 12690. https://doi.org/10.3390/ijms232012690
Rajlic S, Surmann L, Zimmermann P, Weisheit CK, Bindila L, Treede H, Velten M, Daiber A, Duerr GD. Fatty Acid Amide Hydrolase Deficiency Is Associated with Deleterious Cardiac Effects after Myocardial Ischemia and Reperfusion in Mice. International Journal of Molecular Sciences. 2022; 23(20):12690. https://doi.org/10.3390/ijms232012690
Chicago/Turabian StyleRajlic, Sanela, Luise Surmann, Pia Zimmermann, Christina Katharina Weisheit, Laura Bindila, Hendrik Treede, Markus Velten, Andreas Daiber, and Georg Daniel Duerr. 2022. "Fatty Acid Amide Hydrolase Deficiency Is Associated with Deleterious Cardiac Effects after Myocardial Ischemia and Reperfusion in Mice" International Journal of Molecular Sciences 23, no. 20: 12690. https://doi.org/10.3390/ijms232012690