
Non-Reproducibility of Oral Rotenone as a Model for Parkinson’s Disease in Mice

Abstract
:1. Introduction
2. Results
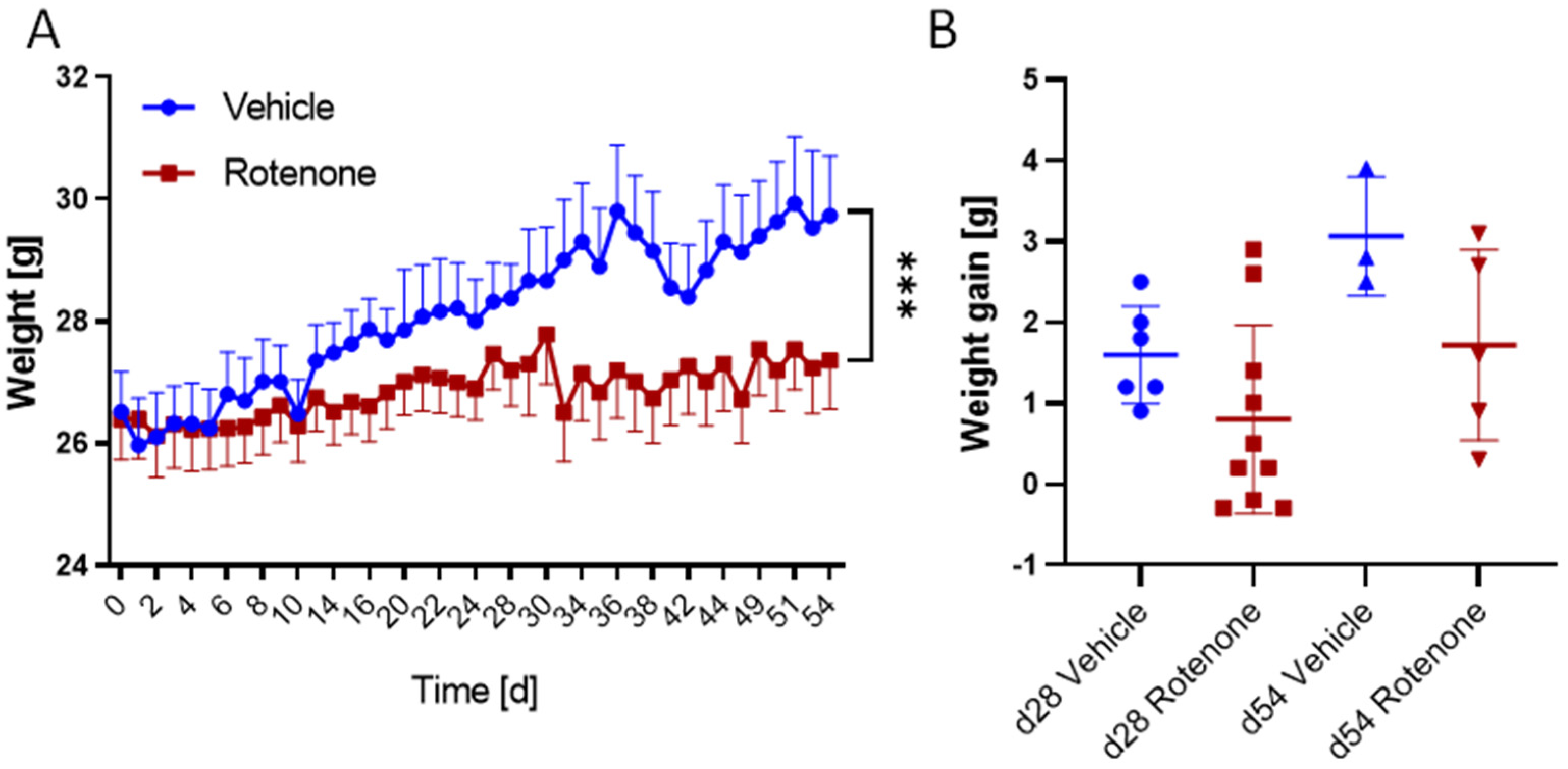
2.1. Weight Gain and Health in Rotenone and Vehicle Group
2.2. Motor Function
2.3. Immunofluorescence
2.4. Rotenone Plasma Concentrations
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Reagents
4.3. Animal Treatment
4.4. RotaRod Test
4.5. Determination of Rotenone Plasma Concentrations
4.6. Immunofluorescence
4.7. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichmann, H.; Brandt, M.D.; Klingelhoefer, L. The nonmotor features of Parkinson’s disease: Pathophysiology and management advances. Curr. Opin. Neurol 2016, 29, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Uversky, V.N.; Fink, A.L. Conformational behavior of human alpha-synuclein is modulated by familial Parkinson’s disease point mutations A30P and A53T. Neurotoxicology 2002, 23, 553–567. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Bower, K.; Fink, A.L. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: Implications for Parkinson’s disease. Neurotoxicology 2002, 23, 527–536. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Paleologou, K.E.; Schmid, A.W.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fredenburg, R.A.; Lansbury, P.T., Jr.; Fernandez, C.O.; Eliezer, D.; Zweckstetter, M.; et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 2008, 283, 16895–16905. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [Green Version]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Breydo, L.; Green, R.; Kis, V.; Puska, G.; Lorincz, P.; Perju-Dumbrava, L.; Giera, R.; Pirker, W.; Lutz, M.; et al. Intracellular processing of disease-associated alpha-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol. Dis. 2014, 69, 76–92. [Google Scholar] [CrossRef]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.H.; Becker, D.; Voos, W.; Leuner, K.; Muller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itier, J.M.; Ibanez, P.; Mena, M.A.; Abbas, N.; Cohen-Salmon, C.; Bohme, G.A.; Laville, M.; Pratt, J.; Corti, O.; Pradier, L.; et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 2003, 12, 2277–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bove, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRx J. Am. Soc. Exp. NeuroTherapeutics 2005, 2, 484–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004, 318, 215–224. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.; Schmidt, W.J. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav. Brain Res. 2002, 136, 317–324. [Google Scholar] [CrossRef]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Sherer, T.B.; Kim, J.H.; Betarbet, R.; Greenamyre, J.T. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol. 2003, 179, 9–16. [Google Scholar] [CrossRef]
- Inden, M.; Kitamura, Y.; Abe, M.; Tamaki, A.; Takata, K.; Taniguchi, T. Parkinsonian rotenone mouse model: Reevaluation of long-term administration of rotenone in C57BL/6 mice. Biol. Pharm. Bull. 2011, 34, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inden, M.; Kitamura, Y.; Takahashi, K.; Takata, K.; Ito, N.; Niwa, R.; Funayama, R.; Nishimura, K.; Taniguchi, T.; Honda, T.; et al. Protection against dopaminergic neurodegeneration in Parkinson’s disease-model animals by a modulator of the oxidized form of DJ-1, a wild-type of familial Parkinson’s disease-linked PARK7. J. Pharmacol. Sci. 2011, 117, 189–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parameshwaran, K.; Irwin, M.H.; Steliou, K.; Pinkert, C.A. Protection by an antioxidant of rotenone-induced neuromotor decline, reactive oxygen species generation and cellular stress in mouse brain. Pharmacol. Biochem. Behav. 2012, 101, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, S.; Ezzat, W.; Elshaer, R.E.; Abd El-Lateef, A.S.; Mohammad, H.M.F.; Elkazaz, A.Y.; Toraih, E.; Zaitone, S.A. Standarized Tribulus terrestris extract protects against rotenone-induced oxidative damage and nigral dopamine neuronal loss in mice. J. Physiol. Pharmacol. 2018, 69, 979–994. [Google Scholar] [CrossRef]
- Chen, Y.; Hou, Y.; Ge, R.; Han, J.; Xu, J.; Chen, J.; Wang, H. Protective effect of roscovitine against rotenone-induced parkinsonism. Restor. Neurol Neurosci. 2018, 36, 629–638. [Google Scholar] [CrossRef]
- Perez-Pardo, P.; Broersen, L.M.; Kliest, T.; van Wijk, N.; Attali, A.; Garssen, J.; Kraneveld, A.D. Additive Effects of Levodopa and a Neurorestorative Diet in a Mouse Model of Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 237. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh Gobi, V.; Rajasankar, S.; Ramkumar, M.; Dhanalakshmi, C.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M.; Chidambaram, R.; Kalandar, A. Agaricus blazei extract abrogates rotenone-induced dopamine depletion and motor deficits by its anti-oxidative and anti-inflammatory properties in Parkinsonic mice. Nutr. Neurosci. 2018, 21, 657–666. [Google Scholar] [CrossRef]
- Wang, T.; Li, C.; Han, B.; Wang, Z.; Meng, X.; Zhang, L.; He, J.; Fu, F. Neuroprotective effects of Danshensu on rotenone-induced Parkinson’s disease models in vitro and in vivo. BMC Complement Med. Ther. 2020, 20, 20. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Li, F.; Ning, J.; Peng, R.; Shang, J.; Liu, H.; Shang, M.; Bao, X.Q.; Zhang, D. Novel compound FLZ alleviates rotenone-induced PD mouse model by suppressing TLR4/MyD88/NF-κB pathway through microbiota-gut-brain axis. Acta Pharm. Sin. B 2021, 11, 2859–2879. [Google Scholar] [CrossRef]
- Pan-Montojo, F.J.; Funk, R.H. Oral administration of rotenone using a gavage and image analysis of alpha-synuclein inclusions in the enteric nervous system. J. Vis. Exp. 2010, 44, e2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasselli, M.; Chaumette, T.; Paillusson, S.; Monnet, Y.; Lafoux, A.; Huchet-Cadiou, C.; Aubert, P.; Hunot, S.; Derkinderen, P.; Neunlist, M. Effects of oral administration of rotenone on gastrointestinal functions in mice. Neurogastroenterol. Motil. 2013, 25, e183–e193. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qian, Y.; Xu, S.; Song, Y.; Xiao, Q. Longitudinal Analysis of Fecal Microbiome and Pathologic Processes in a Rotenone Induced Mice Model of Parkinson’s Disease. Front Aging Neurosci. 2017, 9, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, L.H.; Hara, D.B.; Bicca, M.A.; Poli, A.; Takahashi, R.N. Early signs of colonic inflammation, intestinal dysfunction, and olfactory impairments in the rotenone-induced mouse model of Parkinson’s disease. Behav. Pharmacol. 2018, 29, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, Y.; Si, J.; Pu, M.; Ross, O.A.; McLean, P.J.; Till, L.; Moor, W.; Grover, M.; Kandimalla, K.K.; Margolis, K.G.; et al. Role of gut microbiota in regulating gastrointestinal dysfunction and motor symptoms in a mouse model of Parkinson’s disease. Gut Microbes 2021, 13, 1866974. [Google Scholar] [CrossRef]
- Dodiya, H.B.; Forsyth, C.B.; Voigt, R.M.; Engen, P.A.; Patel, J.; Shaikh, M.; Green, S.J.; Naqib, A.; Roy, A.; Kordower, J.H.; et al. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol. Dis. 2020, 135, 104352. [Google Scholar] [CrossRef]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef]
- Takeuchi, H.; Yanagida, T.; Inden, M.; Takata, K.; Kitamura, Y.; Yamakawa, K.; Sawada, H.; Izumi, Y.; Yamamoto, N.; Kihara, T.; et al. Nicotinic receptor stimulation protects nigral dopaminergic neurons in rotenone-induced Parkinson’s disease models. J. Neurosci. Res. 2009, 87, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.F.; Ho, P.W.; Leung, G.C.; Lam, C.S.; Pang, S.Y.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Combined LRRK2 mutation, aging and chronic low dose oral rotenone as a model of Parkinson’s disease. Sci. Rep. 2017, 7, 40887. [Google Scholar] [CrossRef] [Green Version]
- George, S.; Mok, S.S.; Nurjono, M.; Ayton, S.; Finkelstein, D.I.; Masters, C.L.; Li, Q.X.; Culvenor, J.G. alpha-Synuclein transgenic mice reveal compensatory increases in Parkinson’s disease-associated proteins DJ-1 and parkin and have enhanced alpha-synuclein and PINK1 levels after rotenone treatment. J. Mol. Neurosci. 2010, 42, 243–254. [Google Scholar] [CrossRef]
- Rojo, A.I.; Cavada, C.; de Sagarra, M.R.; Cuadrado, A. Chronic inhalation of rotenone or paraquat does not induce Parkinson’s disease symptoms in mice or rats. Exp. Neurol. 2007, 208, 120–126. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.D.; Song, L.K.; Li, J.; Chu, S.F.; Yuan, Y.H.; Chen, N.H. Environment-contact administration of rotenone: A new rodent model of Parkinson’s disease. Behav. Brain Res. 2015, 294, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Kang, S.S.; Liu, X.; Chen, G.; Zhang, Z.; Chandrasekharan, B.; Alam, A.M.; Neish, A.S.; Cao, X.; Ye, K. Initiation of Parkinson’s disease from gut to brain by δ-secretase. Cell Res. 2020, 30, 70–87. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Lu, Y.; Tian, H.; Duan, C.; Lu, L.; Gao, G.; Wu, X.; Wang, X.; Yang, H. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models. Autophagy 2018, 14, 845–861. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Lin, H.Y.; Ho, E.P.; Ke, Y.C.; Cheng, M.F.; Shiue, C.Y.; Wu, C.H.; Liao, P.H.; Hsu, A.Y.; Chu, L.A.; et al. Mild Chronic Colitis Triggers Parkinsonism in LRRK2 Mutant Mice Through Activating TNF-α Pathway. Mov. Disord. 2022, 37, 745–757. [Google Scholar] [CrossRef]
- Innos, J.; Hickey, M.A. Using Rotenone to Model Parkinson’s Disease in Mice: A Review of the Role of Pharmacokinetics. Chem. Res. Toxicol. 2021, 34, 1223–1239. [Google Scholar] [CrossRef]
- Ulusoy, A.; Rusconi, R.; Perez-Revuelta, B.I.; Musgrove, R.E.; Helwig, M.; Winzen-Reichert, B.; Di Monte, D.A. Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol. Med. 2013, 5, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, A.; Musgrove, R.E.; Rusconi, R.; Klinkenberg, M.; Helwig, M.; Schneider, A.; Di Monte, D.A. Neuron-to-neuron alpha-synuclein propagation in vivo is independent of neuronal injury. Acta Neuropathol. Commun. 2015, 3, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusconi, R.; Ulusoy, A.; Aboutalebi, H.; Di Monte, D.A. Long-lasting pathological consequences of overexpression-induced alpha-synuclein spreading in the rat brain. Aging Cell 2018, 17, e12727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.M.; Alsahaf, H.; Streete, P.; Dargan, P.I.; Jones, A.L. Fatality after deliberate ingestion of the pesticide rotenone: A case report. Crit. Care 2005, 9, R280–R284. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huo, M.; Jin, Y.; Wang, Y.; Wang, X.; Yu, W.; Jiang, X. Rotenone induces hepatotoxicity in rats by activating the mitochondrial pathway of apoptosis. Toxicol. Mech. Methods 2022, 32, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Bjorklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Manfredsson, F.P.; Luk, K.C.; Benskey, M.J.; Gezer, A.; Garcia, J.; Kuhn, N.C.; Sandoval, I.M.; Patterson, J.R.; O’Mara, A.; Yonkers, R.; et al. Induction of alpha-synuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol. Dis. 2018, 112, 106–118. [Google Scholar] [CrossRef]
- Abdo, K.M.; Eustis, S.L.; Haseman, J.; Huff, J.E.; Peters, A.; Persing, R. Toxicity and carcinogenicity of rotenone given in the feed to F344/N rats and B6C3F1 mice for up to two years. Drug Chem. Toxicol. 1988, 11, 225–235. [Google Scholar] [CrossRef]
- Kumar, R.; Jangir, D.K.; Verma, G.; Shekhar, S.; Hanpude, P.; Kumar, S.; Kumari, R.; Singh, N.; Sarovar Bhavesh, N.; Ranjan Jana, N.; et al. S-nitrosylation of UCHL1 induces its structural instability and promotes alpha-synuclein aggregation. Sci. Rep. 2017, 7, 44558. [Google Scholar] [CrossRef]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Wang, W.; Chen, C.; Tang, L.; Liang, Y.; Zhang, Z.; Lu, Y.; Zhao, Y. UHPLC-MS-based metabolomics and chemoinformatics study reveals the neuroprotective effect and chemical characteristic in Parkinson’s disease mice after oral administration of Wen-Shen-Yang-Gan decoction. Aging 2021, 13, 19510–19528. [Google Scholar] [CrossRef]
- Khairnar, A.; Ruda-Kucerova, J.; Arab, A.; Hadjistyllis, C.; Sejnoha Minsterova, A.; Shang, Q.; Chovsepian, A.; Drazanova, E.; Szabó, N.; Starcuk, Z., Jr.; et al. Diffusion kurtosis imaging detects the time-dependent progress of pathological changes in the oral rotenone mouse model of Parkinson’s disease. J. Neurochem. 2021, 158, 779–797. [Google Scholar] [CrossRef]
- Jain, J.; Hasan, W.; Biswas, P.; Yadav, R.S.; Jat, D. Neuroprotective effect of quercetin against rotenone-induced neuroinflammation and alterations in mice behavior. J. Biochem. Mol. Toxicol. 2022, 36, e23165. [Google Scholar] [CrossRef]
- McQuade, R.M.; Singleton, L.M.; Wu, H.; Lee, S.; Constable, R.; Di Natale, M.; Ringuet, M.T.; Berger, J.P.; Kauhausen, J.; Parish, C.L.; et al. The association of enteric neuropathy with gut phenotypes in acute and progressive models of Parkinson’s disease. Sci. Rep. 2021, 11, 7934. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Scuto, M.; Fusco, R.; Trovato, A.; Ontario, M.L.; Crea, R.; Di Paola, R.; Cuzzocrea, S.; Calabrese, V. Anti-inflammatory and Anti-oxidant Activity of Hidrox((R)) in Rotenone-Induced Parkinson’s Disease in Mice. Antioxidants 2020, 9, 824. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Vehicle Day 28 (n = 6) | Rotenone Day 28 (n = 10) | Vehicle Day 53 (n = 3) | Rotenone Day 54 (n = 5) | |
|---|---|---|---|---|
| Survival [%] | 100 | 100 | 100 | 100 |
| Weight gain [g] Mean ± SD | 1.6 ± 0.6 | 0.8 ± 1.1 | 3.1 ± 0.6 | 1.7 ± 1.1 |
| RotaRod [% BL] Mean ± SD | 99.6 ± 14.6 | 100.6 ± 13.1 | 98.7 ± 20.5 | 96.1 ± 20.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niederberger, E.; Wilken-Schmitz, A.; Manderscheid, C.; Schreiber, Y.; Gurke, R.; Tegeder, I. Non-Reproducibility of Oral Rotenone as a Model for Parkinson’s Disease in Mice. Int. J. Mol. Sci. 2022, 23, 12658. https://doi.org/10.3390/ijms232012658
Niederberger E, Wilken-Schmitz A, Manderscheid C, Schreiber Y, Gurke R, Tegeder I. Non-Reproducibility of Oral Rotenone as a Model for Parkinson’s Disease in Mice. International Journal of Molecular Sciences. 2022; 23(20):12658. https://doi.org/10.3390/ijms232012658
Chicago/Turabian StyleNiederberger, Ellen, Annett Wilken-Schmitz, Christine Manderscheid, Yannick Schreiber, Robert Gurke, and Irmgard Tegeder. 2022. "Non-Reproducibility of Oral Rotenone as a Model for Parkinson’s Disease in Mice" International Journal of Molecular Sciences 23, no. 20: 12658. https://doi.org/10.3390/ijms232012658