
Small Extracellular Vesicles from Hypoxic Triple-Negative Breast Cancer Cells Induce Oxygen-Dependent Cell Invasion
 ,
,  , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
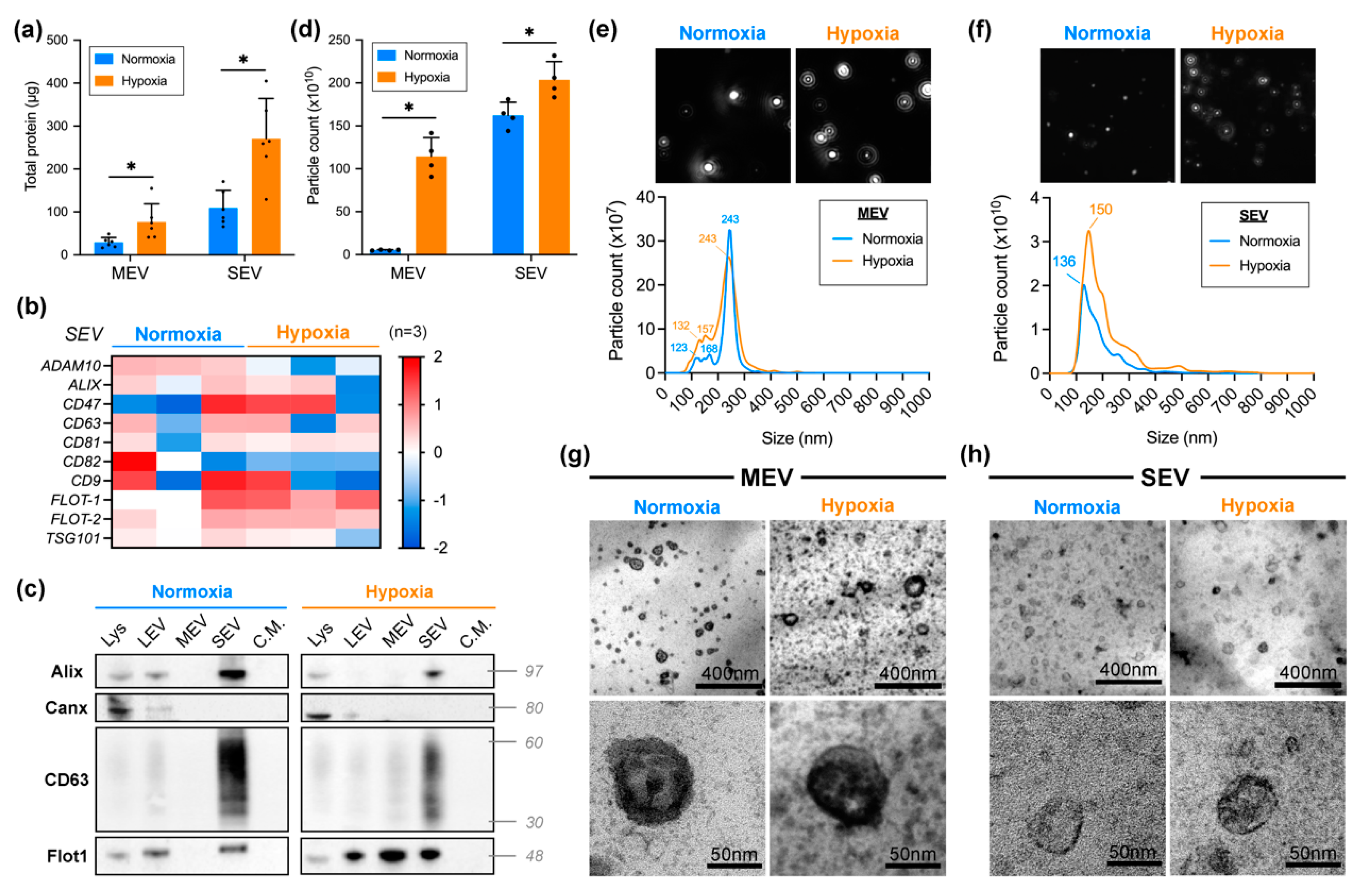
2.1. SEVh Isolation and Characterization
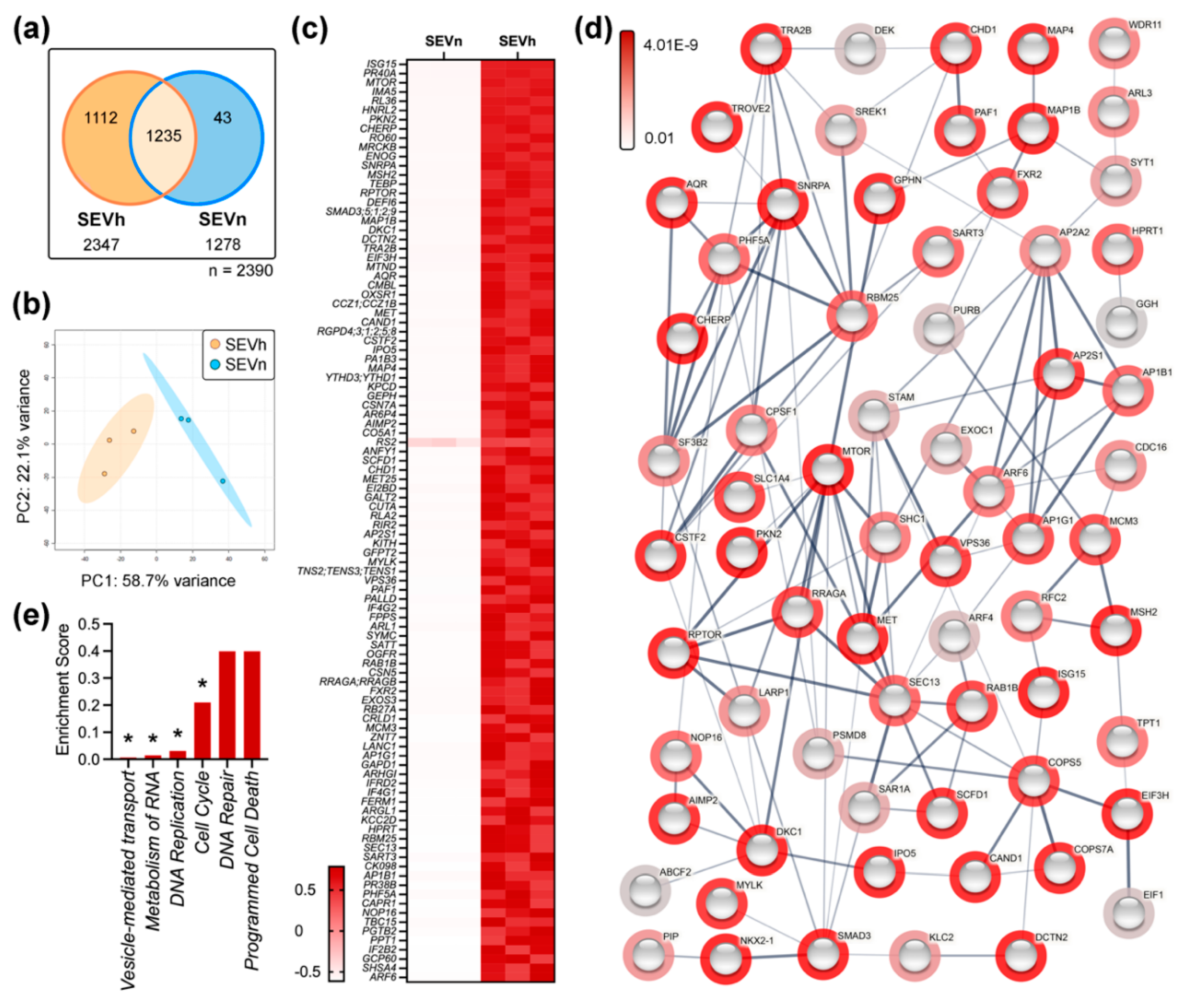
2.2. SEVh Proteome Is Enriched with Components of Pro-Invasive Signaling Pathways
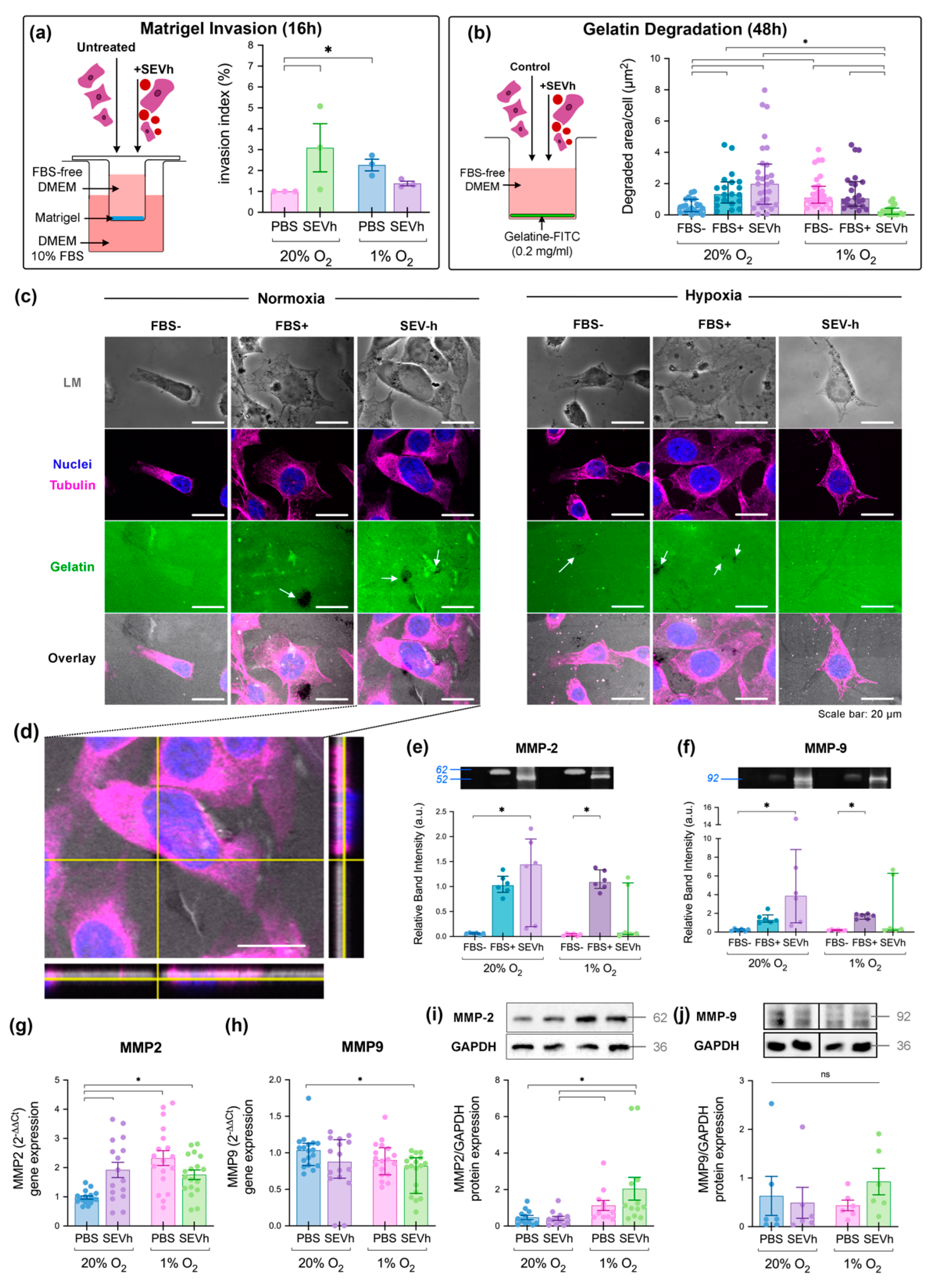
2.3. SEV-h Signaling Triggers Long-Term MDA-MB-231 Invasion
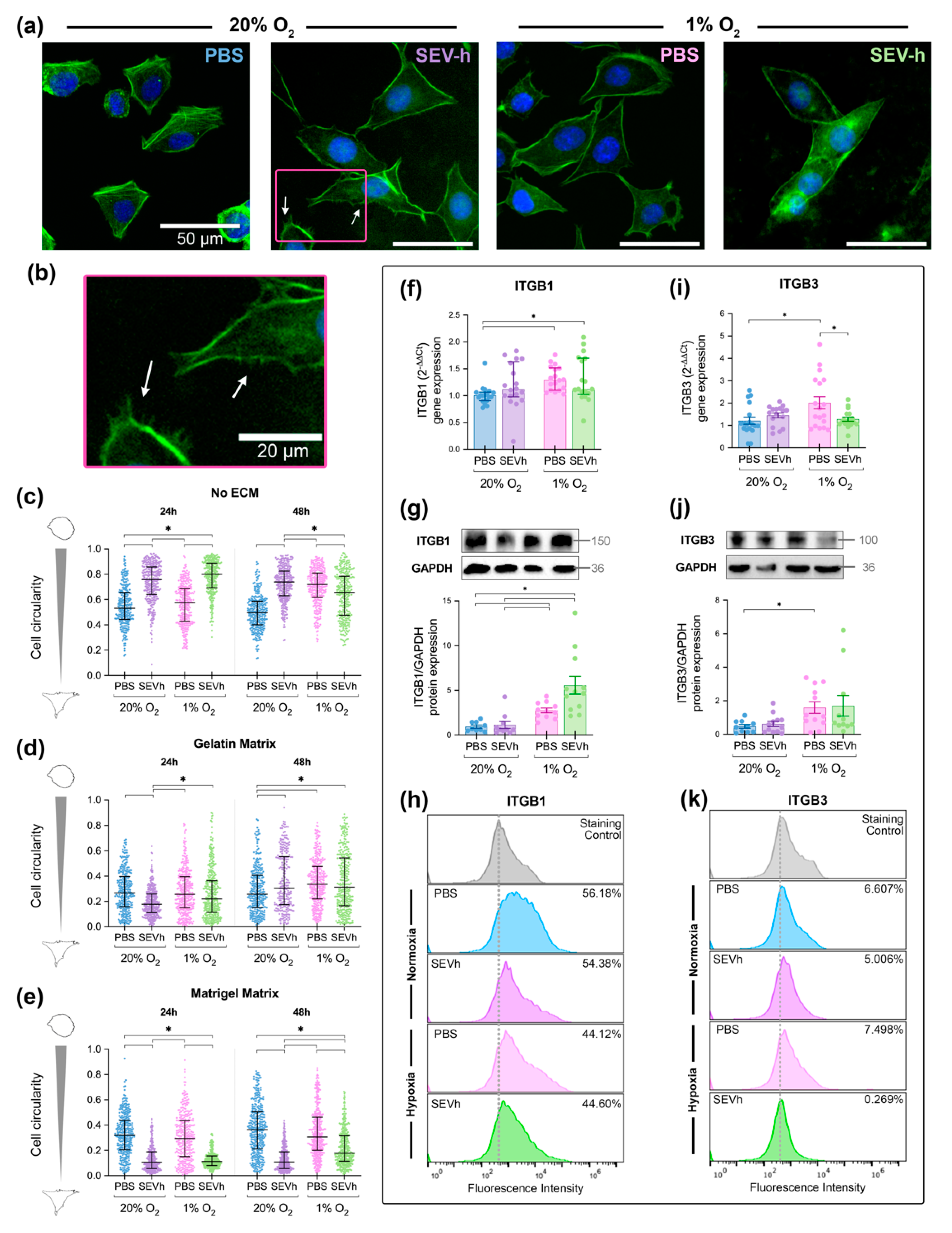
2.4. SEVh Exposure Favors Migratory Morphology of MDA-MB-231 Cells in Normoxia
2.5. Hypoxia Interferes with β1 and β3 Integrin Subunit Expression in TNBC Cells
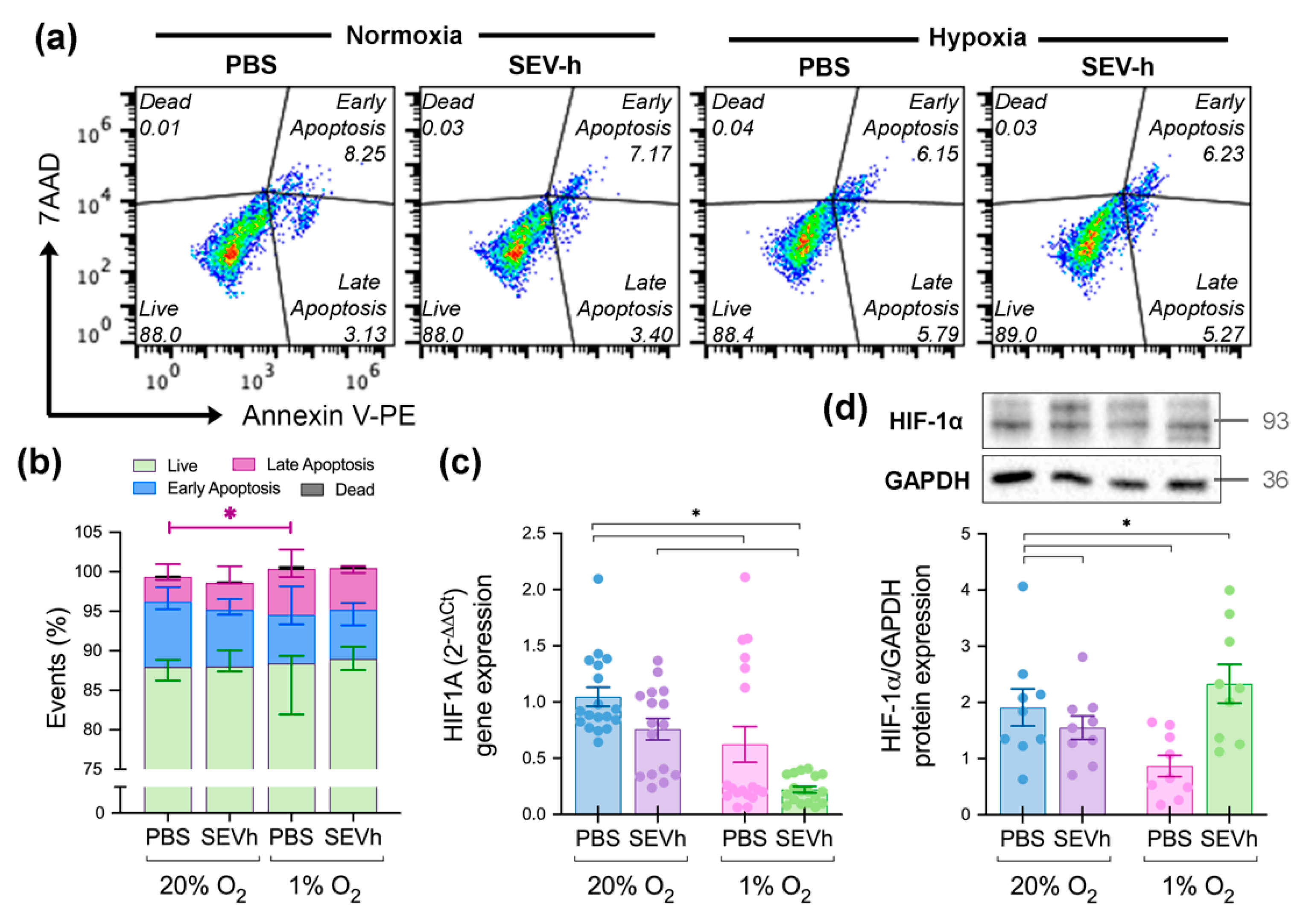
2.6. Hypoxia-Derived Apoptosis Is Modulated by SEVh Signaling
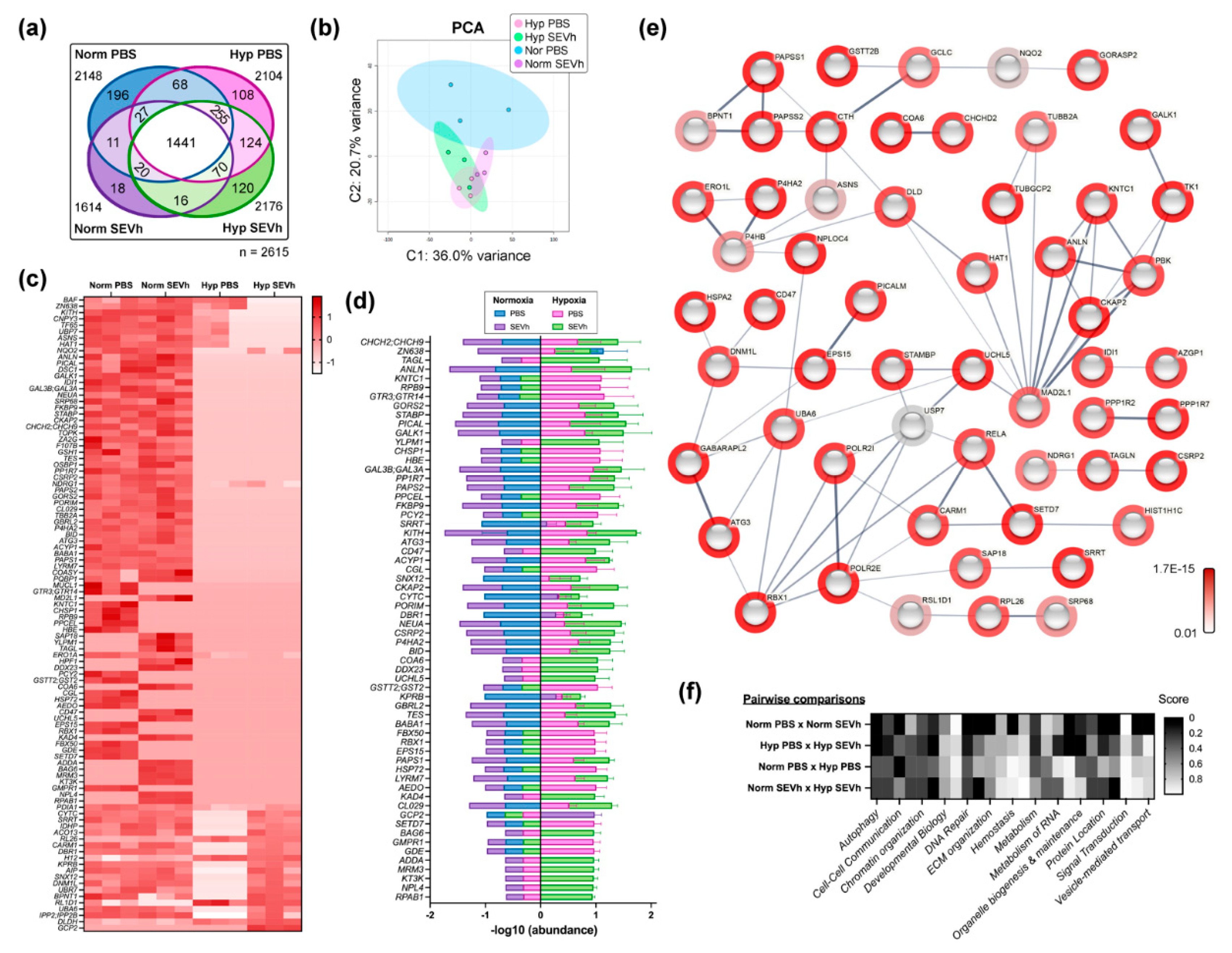
2.7. MDA-MB-231 Comparative Proteome Shows That SEVh Favors a Shift in Cytoskeleton Arrangements and Cell Cycle in Normoxia
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. EV Isolation by Differential Ultracentrifugation
4.3. Nanoparticle Tracking Analysis
4.4. Transmission Electron Microscopy (TEM)
4.5. Protein Quantification
4.6. Experimental Design
4.7. Cell Lysate
4.8. Western Blotting
4.9. Protein Extraction
4.10. Label-Free LC-MS/MS Proteomics
4.11. Bioinformatic Analysis and Data Enrichment
4.12. Gelatin Zymography
4.13. Integrin Immunophenotyping by Flow Cytometry
4.14. Matrigel Invasion Assay
4.15. Fluorescent Gelatin Degradation Assay
4.16. Cell Morphology Assay
4.17. Apoptosis Assay
4.18. Total RNA Extraction and cDNA Synthesis
4.19. Gene Expression by RT-qPCR
4.20. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, A.; Deep, G. Hypoxia in Tumor Microenvironment Regulates Exosome Biogenesis: Molecular Mechanisms and Translational Opportunities. Cancer Lett. 2020, 479, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ebright, R.Y.; Zachariah, M.A.; Micalizzi, D.S.; Wittner, B.S.; Niederhoffer, K.L.; Nieman, L.T.; Chirn, B.; Wiley, D.F.; Wesley, B.; Shaw, B.; et al. HIF1A Signaling Selectively Supports Proliferation of Breast Cancer in the Brain. Nat. Commun. 2020, 11, 6311. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Mao, X.; Brown-Clay, J.; Moreau, F.; al Absi, A.; Wurzer, H.; Sousa, B.; Schmitt, F.; Berchem, G.; Janji, B.; et al. Hypoxia Promotes Breast Cancer Cell Invasion through HIF-1α-Mediated up-Regulation of the Invadopodial Actin Bundling Protein CSRP2. Sci. Rep. 2018, 8, 10191. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Xiao, Y.; Zhu, X.; Ning, Z.; Xu, H.; Wu, H. Hypoxia Regulates Stemness of Breast Cancer MDA-MB-231 Cells. Med. Oncol. 2016, 33, 42. [Google Scholar] [CrossRef] [Green Version]
- Begg, K.; Tavassoli, M. Inside the Hypoxic Tumour: Reprogramming of the DDR and Radioresistance. Cell Death Discov. 2020, 6, 77. [Google Scholar] [CrossRef]
- McAleese, C.E.; Choudhury, C.; Butcher, N.J.; Minchin, R.F. Hypoxia-Mediated Drug Resistance in Breast Cancers. Cancer Lett. 2021, 502, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic Enhancement of Exosome Release by Breast Cancer Cells. BMC Cancer 2012, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and Directions in Studying Cell–Cell Communication by Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; di Giannatale, A.; Ceder, S.; et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzolo, G.; Albanese, N.N.; di Cara, G.; Gygax, D.; Vittorelli, M.L.; Pucci-Minafra, I. Proteomic Analysis of Exosome-like Vesicles Derived from Breast Cancer Cells. Anticancer Res. 2012, 32, 847–860. [Google Scholar] [PubMed]
- Jiang, H.; Zhao, H.; Zhang, M.; He, Y.; Li, X.; Xu, Y.; Liu, X. Hypoxia Induced Changes of Exosome Cargo and Subsequent Biological Effects. Front. Immunol. 2022, 13, 824188. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia 2015, 2015, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Bister, N.; Pistono, C.; Huremagic, B.; Jolkkonen, J.; Giugno, R.; Malm, T. Hypoxia and Extracellular Vesicles: A Review on Methods, Vesicular Cargo and Functions. J. Extracell Vesicles 2020, 10, e12002. [Google Scholar] [CrossRef]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of Breast Cancer Cells Mediated by Transforming Growth Factor-β in Exosomes from Cancer Cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-Inducible Factors and RAB22A Mediate Formation of Microvesicles That Stimulate Breast Cancer Invasion and Metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.O.; Jo, H.; Yu, J.H.; Gambhir, S.S.; Pratx, G. Development and MPI Tracking of Novel Hypoxia-Targeted Theranostic Exosomes. Biomaterials 2018, 177, 139–148. [Google Scholar] [CrossRef]
- Yousefi, H.; Vatanmakanian, M.; Mahdiannasser, M.; Mashouri, L.; Alahari, N.v.; Monjezi, M.R.; Ilbeigi, S.; Alahari, S.K. Understanding the Role of Integrins in Breast Cancer Invasion, Metastasis, Angiogenesis, and Drug Resistance. Oncogene 2021, 40, 1043–1063. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, Microtubules, and Vimentin Intermediate Filaments Cooperate for Elongation of Invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, D.; Kirkbride, K.C.; Costello, K.; Clark, E.S.; Sinha, S.; Grega-Larson, N.; Tyska, M.J.; Weaver, A.M. Exosome Secretion Is Enhanced by Invadopodia and Drives Invasive Behavior. Cell Rep. 2013, 5, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeown, S.R. Defining Normoxia, Physoxia and Hypoxia in Tumours—Implications for Treatment Response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- van Deun, J.; Mestdagh, P.; Agostinis, P.; Akay, Ö.; Anand, S.; Anckaert, J.; Martinez, Z.A.; Baetens, T.; Beghein, E.; Bertier, L.; et al. EV-TRACK: Transparent Reporting and Centralizing Knowledge in Extracellular Vesicle Research. Nat. Methods 2017, 14, 228–232. [Google Scholar] [PubMed]
- Hermann, M.R.; Jakobson, M.; Colo, G.P.; Rognoni, E.; Jakobson, M.; Kupatt, C.; Posern, G.; Fässler, R. Integrins Synergise to Induce Expression of the MRTF-A-SRF Target Gene ISG15 for Promoting Cancer Cell Invasion. J. Cell Sci. 2016, 129, 1391–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossmann, D.; Park, S.; Hall, M.N. MTOR Signalling and Cellular Metabolism Are Mutual Determinants in Cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-Dependent and Smad-Independent Pathways in TGF-b Family Signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Gaudet, P.; Livstone, M.S.; Lewis, S.E.; Thomas, P.D. Phylogenetic-Based Propagation of Functional Annotations within the Gene Ontology Consortium. Brief. Bioinform. 2011, 12, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Om Alblazi, K.M.; Siar, C.H. Cellular Protrusions—Lamellipodia, Filopodia, Invadopodia and Podosomes- and Their Roles in Progression of Orofacial Tumours: Current Understanding. Asian Pac. J. Cancer Prev. 2015, 16, 2187–2191. [Google Scholar] [CrossRef]
- Zhang, X.; Chan, T.; Mak, M. Morphodynamic Signatures of MDA-MB-231 Single Cells and Cell Doublets Undergoing Invasion in Confined Microenvironments. Sci. Rep. 2021, 11, 6529. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Li, P.; Ji, C. Cell Death Conversion under Hypoxic Condition in Tumor Development and Therapy. Int. J. Mol. Sci. 2015, 16, 25536–25551. [Google Scholar] [CrossRef] [Green Version]
- Greijer, A.E.; van der Wall, E. The Role of Hypoxia Inducible Factor 1 (HIF-1) in Hypoxia Induced Apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Werb, Z. Invasion and Metastasis—Recent Advances and Future Challenges. J. Mol. Med. 2015, 93, 361–368. [Google Scholar] [CrossRef]
- Taware, R.; Taunk, K.; Kumar, T.V.S.; Pereira, J.A.M.; Câmara, J.S.; Nagarajaram, H.A.; Kundu, G.C.; Rapole, S. Extracellular Volatilomic Alterations Induced by Hypoxia in Breast Cancer Cells. Metabolomics 2020, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Singh, A.; Mattheolabakis, G.; Suresh, M.; Amiji, M.M. Exosome Mediated Communication within the Tumor Microenvironment. J. Control. Release 2015, 219, 278–294. [Google Scholar] [CrossRef]
- Sesé, M.; Fuentes, P.; Esteve-Codina, A.; Béjar, E.; McGrail, K.; Thomas, G.; Aasen, T.; Ramón y Cajal, S. Hypoxia-Mediated Translational Activation of ITGB3 in Breast Cancer Cells Enhances TGF-β Signaling and Malignant Features in Vitro and in Vivo. Oncotarget 2017, 8, 114856–114876. [Google Scholar] [CrossRef] [Green Version]
- Grayson, W.L.; Zhao, F.; Bunnell, B.; Ma, T. Hypoxia Enhances Proliferation and Tissue Formation of Human Mesenchymal Stem Cells. Biochem. Biophys. Res. Commun. 2007, 358, 948–953. [Google Scholar] [CrossRef]
- Schmaltz, C.; Hardenbergh, P.H.; Wells, A.; Fisher, D.E. Regulation of Proliferation-Survival Decisions during Tumor Cell Hypoxia. Mol. Cell Biol. 1998, 18, 2845–2854. [Google Scholar] [CrossRef]
- Bernhardt, S.; Tönsing, C.; Mitra, D.; Erdem, N.; Müller-Decker, K.; Korf, U.; Kreutz, C.; Timmer, J.; Wiemann, S. Functional Proteomics of Breast Cancer Metabolism Identifies GLUL as Responder during Hypoxic Adaptation. J. Proteome Res. 2019, 18, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Hannafon, B.N.; Gin, A.L.; Xu, Y.; Bruns, M.; Calloway, C.L.; Ding, W. Metastasis-Associated Protein 1 (MTA1) Is Transferred by Exosomes and Contributes to the Regulation of Hypoxia and Estrogen Signaling in Breast Cancer Cells. Cell Commun. Signal. 2019, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casali, B.C.; Gozzer, L.T.; Baptista, M.P.; Altei, W.F.; Selistre-De-araújo, H.S. The Effects of Avβ3 Integrin Blockage in Breast Tumor and Endothelial Cells under Hypoxia In Vitro. Int. J. Mol. Sci. 2022, 23, 1745. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; van der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; van Diest, P.J.; van der Wall, E. Levels of Hypoxia-Inducible Factor-1α Independently Predict Prognosis in Patients with Lymph Node Negative Breast Carcinoma. Cancer 2003, 97, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Rana, N.K.; Singh, P.; Koch, B. CoCl2 Simulated Hypoxia Induce Cell Proliferation and Alter the Expression Pattern of Hypoxia Associated Genes Involved in Angiogenesis and Apoptosis. Biol. Res. 2019, 52, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Rashid, M.; Zadeh, L.R.; Baradaran, B.; Molavi, O.; Ghesmati, Z.; Sabzichi, M.; Ramezani, F. Up-down Regulation of HIF-1α in Cancer Progression. Gene 2021, 798, 145796. [Google Scholar] [CrossRef]
- Stegen, S.; Laperre, K.; Eelen, G.; Rinaldi, G.; Fraisl, P.; Torrekens, S.; van Looveren, R.; Loopmans, S.; Bultynck, G.; Vinckier, S.; et al. HIF-1α Metabolically Controls Collagen Synthesis and Modification in Chondrocytes. Nature 2019, 565, 511–515. [Google Scholar] [CrossRef]
- Ito, K.; Kitajima, Y.; Kai, K.; Matsufuji, S.; Yamada, K.; Egawa, N.; Kitagawa, H.; Okuyama, K.; Tanaka, T.; Noshiro, H. Matrix Metalloproteinase-1 Expression Is Regulated by HIF-1-dependent and Epigenetic Mechanisms and Serves a Tumor-suppressive Role in Gastric Cancer Progression. Int. J. Oncol. 2021, 59, 102. [Google Scholar] [CrossRef]
- Overall, C.M.; López-Otín, C. Strategies for MMP Inhibition in Cancer: Innovations for the Post-Trial Era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef]
- Cui, H.; Grosso, S.; Schelter, F.; Mari, B.; Krüger, A. On the Pro-Metastatic Stress Response to Cancer Therapies: Evidence for a Positive Co-Operation between TIMP-1, HIF-1α, and MiR-210. Front. Pharmacol. 2012, 3, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholami, A.M.; Hahne, H.; Wu, Z.; Auer, F.J.; Meng, C.; Wilhelm, M.; Kuster, B. Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Rep. 2013, 4, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, S.N.; Meckes, D.G. Extracellular Vesicle Integrins Distinguish Unique Cancers. Proteomes 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte, M.A.; Nolte-’t Hoen, E.N.M.; Margadant, C. Integrins Control Vesicular Trafficking; New Tricks for Old Dogs. Trends Biochem. Sci. 2021, 46, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Ford, S.; Yan, C.; Chung, J. Roles of Integrins in Regulating Metastatic Potentials of Cancer Cell Derived Exosomes. Mol. Cell Toxicol. 2019, 15, 233–237. [Google Scholar] [CrossRef]
- Montenegro, C.F.; Casali, B.C.; Lino, R.L.B.; Pachane, B.C.; Santos, P.K.; Horwitz, A.R.; Selistre-De-Araujo, H.S.; Lamers, M.L. Inhibition of Avβ3 Integrin Induces Loss of Cell Directionality of Oral Squamous Carcinoma Cells (OSCC). PLoS ONE 2017, 12, e0176226. [Google Scholar] [CrossRef] [Green Version]
- DeRita, R.M.; Sayeed, A.; Garcia, V.; Krishn, S.R.; Shields, C.D.; Sarker, S.; Friedman, A.; McCue, P.; Molugu, S.K.; Rodeck, U.; et al. Tumor-Derived Extracellular Vesicles Require Β1 Integrins to Promote Anchorage-Independent Growth. iScience 2019, 14, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Carter, R.Z.; Micocci, K.C.; Natoli, A.; Redvers, R.P.; Paquet-Fifield, S.; Martin, A.C.B.M.; Denoyer, D.; Ling, X.; Kim, S.H.; Tomasin, R.; et al. Tumour but Not Stromal Expression of β 3 Integrin Is Essential, and Is Required Early, for Spontaneous Dissemination of Bone-Metastatic Breast Cancer. J. Pathol. 2015, 235, 760–772. [Google Scholar] [CrossRef]
- Moritz, M.N.O.; Merkel, A.R.; Feldman, E.G.; Selistre-de-Araujo, H.S.; Rhoades (Sterling), J.A. Biphasic A2β1 Integrin Expression in Breast Cancer Metastasis to Bone. Int. J. Mol. Sci. 2021, 22, 6906. [Google Scholar] [CrossRef]
- dos Santos, P.K.; Altei, W.F.; Danilucci, T.M.; Lino, R.L.B.; Pachane, B.C.; Nunes, A.C.C.; Selistre-de-Araujo, H.S. Alternagin-C (ALT-C), a Disintegrin-like Protein, Attenuates Alpha2beta1 Integrin and VEGF Receptor 2 Signaling Resulting in Angiogenesis Inhibition. Biochimie 2020, 174, 144–158. [Google Scholar] [CrossRef]
- Danilucci, T.M.; Santos, P.K.; Pachane, B.C.; Pisani, G.F.D.; Lino, R.L.B.; Casali, B.C.; Altei, W.F.; Selistre-de-Araujo, H.S. Recombinant RGD-Disintegrin DisBa-01 Blocks Integrin Avβ3 and Impairs VEGF Signaling in Endothelial Cells. Cell Commun. Signal. 2019, 17, 27. [Google Scholar] [CrossRef] [Green Version]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The Final Steps of Integrin Activation: The End Game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, F.; Rüdiger, D.; Zahler, S.; Engelke, H. Fiber Stiffness, Pore Size and Adhesion Control Migratory Phenotype of MDA-MB-231 Cells in Collagen Gels. PLoS ONE 2019, 14, e0225215. [Google Scholar] [CrossRef] [PubMed]
- Strande, V.; Canelle, L.; Tastet, C.; Burlet-Schiltz, O.; Monsarrat, B.; Hondermarck, H. The Proteome of the Human Breast Cancer Cell Line MDA-MB-231: Analysis by LTQ-Orbitrap Mass Spectrometry. Proteomics Clin. Appl. 2009, 3, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of Oxygen Delivery to Cells in Culture: An Underappreciated Problem in Basic and Translational Research. Free Radic. Biol. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carrera, I.; Frise, E.; Verena, K.; Mark, L.; Tobias, P.; Stephan, P.; Curtis, R.; Stephan, S.; Benjamin, S.; et al. Fiji: An Open Platform for Biological Image Analysis. Nat. Methods 2009, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [Green Version]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A. UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. ReactomeGSA—Efficient Multi-Omics Comparative Pathway Analysis. Mol. Cell. Proteom. 2020, 19, 2115–2124. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Leber, T.M.; Balkwill, F.R. Zymography: A Single-Step Staining Method for Quantitation of Proteolytic Activity on Substrate Gels. Anal. Biochem. 1997, 249, 24–28. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pachane, B.C.; Nunes, A.C.C.; Cataldi, T.R.; Micocci, K.C.; Moreira, B.C.; Labate, C.A.; Selistre-de-Araujo, H.S.; Altei, W.F. Small Extracellular Vesicles from Hypoxic Triple-Negative Breast Cancer Cells Induce Oxygen-Dependent Cell Invasion. Int. J. Mol. Sci. 2022, 23, 12646. https://doi.org/10.3390/ijms232012646
Pachane BC, Nunes ACC, Cataldi TR, Micocci KC, Moreira BC, Labate CA, Selistre-de-Araujo HS, Altei WF. Small Extracellular Vesicles from Hypoxic Triple-Negative Breast Cancer Cells Induce Oxygen-Dependent Cell Invasion. International Journal of Molecular Sciences. 2022; 23(20):12646. https://doi.org/10.3390/ijms232012646
Chicago/Turabian StylePachane, Bianca Cruz, Ana Carolina Caetano Nunes, Thais Regiani Cataldi, Kelli Cristina Micocci, Bianca Caruso Moreira, Carlos Alberto Labate, Heloisa Sobreiro Selistre-de-Araujo, and Wanessa Fernanda Altei. 2022. "Small Extracellular Vesicles from Hypoxic Triple-Negative Breast Cancer Cells Induce Oxygen-Dependent Cell Invasion" International Journal of Molecular Sciences 23, no. 20: 12646. https://doi.org/10.3390/ijms232012646