
Selective and Reversible 1,3-Dipolar Cycloaddition of 2-(2-Oxoindoline-3-ylidene)acetates with Nitrones in the Synthesis of Functionalized Spiroisoxazolidines
 , , , and
, , , and
Abstract
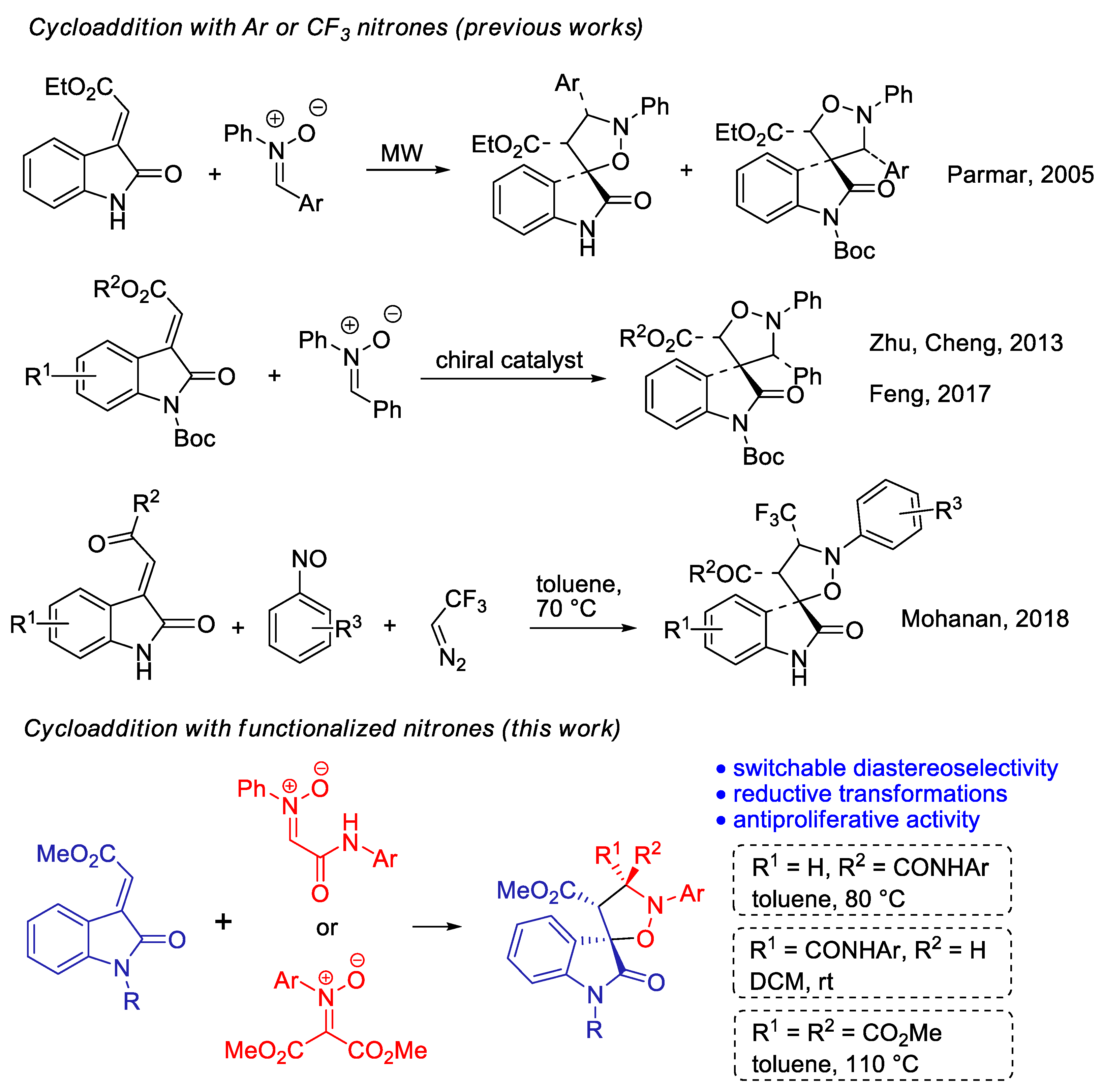
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Structural Characterization
2.2. Transformations of the Cycloadducts
2.3. Antiproliferative Activity
3. Materials and Methods
3.1. General Information
3.2. Synthetic Methods and Analytic Data of Compounds
3.2.1. General Procedure for Obtaining Cycloadducts 3, 3′, and 4
3.2.2. General Procedure for Obtaining Cycloadducts 3′
3.2.3. Procedure for Obtaining Cycloadduct 3′e
3.2.4. General Procedure for Obtaining Cycloadducts 6
3.2.5. General Procedure for Obtaining 1,3-Aminoalcohols 7 and 7′
3.2.6. Procedure for Obtaining Lactone 8
3.3. Bioactivity Assay
3.3.1. Cell Culture
3.3.2. Antiproliferative Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintavalla, A. Spirolactones: Recent Advances in Natural Products, Bioactive Compounds and Synthetic Strategies. Curr. Med. Chem. 2018, 25, 917–962. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183. [Google Scholar] [CrossRef]
- Chupakhin, E.; Babich, O.; Prosekov, A.; Asyakina, L.; Krasavin, M. Spirocyclic Motifs in Natural Products. Molecules 2019, 24, 4165. [Google Scholar] [CrossRef] [Green Version]
- Saraswat, P.; Jeyabalan, G.; Hassan, M.Z.; Rahman, M.U.; Nyola, N.K. Review of synthesis and various biological activities of spiro heterocyclic compounds comprising oxindole and pyrrolidine moities. Synth. Commun. 2016, 46, 1643–1664. [Google Scholar] [CrossRef]
- Molteni, G.; Silvani, A. Spiro-2-oxindoles via 1,3-dipolar cycloadditions. A decade update. Eur. J. Org. Chem. 2021, 11, 1653–1675. [Google Scholar] [CrossRef]
- Zhou, L.-M.; Yang, G.-F. An overview of spirooxindole as a promising scaffold for novel drug discovery. Expert Opin. Drug Discov. 2020, 15, 603–625. [Google Scholar] [CrossRef]
- Rojas-Duran, R.; González-Aspajo, G.; Ruiz-Martel, C.; Bourdy, G.; Doroteo-Ortega, V.H.; Alban-Castillo, J.; Robert, G.; Auberger, P.; Deharo, E. Anti-inflammatory activity of Mitraphylline isolated from Uncaria tomentosa bark. J. Ethnopharmacol. 2012, 143, 801–804. [Google Scholar] [CrossRef]
- Yu, B.; Yu, D.-Q.; Liu, H.-M. Spirooxindoles: Promising scaffolds for anticancer agents. Eur. J. Med. Chem. 2015, 97, 673–698. [Google Scholar] [CrossRef]
- Chaudhari, P.; Bari, S.; Surana, S.; Shirkhedkar, A.; Wakode, S.; Shelar, S.; Racharla, S.; Ugale, V.; Ghodke, M. Logical synthetic strategies and structure-activity relationship of indolin-2-one hybrids as small molecule anticancer agents: An overview. J. Mol. Struct. 2022, 1247, 131280. [Google Scholar] [CrossRef]
- Kumar, R.; Takkar, P. Repositioning of Isatin hybrids as novel anti-tubercular agents overcoming pre-existing antibiotics resistance. Med. Chem. Res. 2021, 30, 847–876. [Google Scholar] [CrossRef]
- Marti, C.; Carreira, E.M. Construction of Spiro[pyrrolidine-3,3′-oxindoles] − Recent Applications to the Synthesis of Oxindole Alkaloids. Eur. J. Org. Chem. 2003, 2003, 2209–2219. [Google Scholar] [CrossRef]
- Santos, M.M.M. Recent advances in the synthesis of biologically active spirooxindoles. Tetrahedron 2014, 70, 9735–9757. [Google Scholar] [CrossRef]
- Bariwal, J.; Voskressensky, L.G.; Van der Eycken, E.V. Recent advances in spirocyclization of indole derivatives. Chem. Soc. Rev. 2018, 47, 3831–3848. [Google Scholar] [CrossRef] [Green Version]
- Singh, G.S.; Desta, Z.Y. Isatins As Privileged Molecules in Design and Synthesis of Spiro-Fused Cyclic Frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef]
- Saeed, R.; Sakla, A.P.; Shankaraiah, N. An update on the progress of cycloaddition reactions of 3-methyleneindolinones in the past decade: Versatile approaches to spirooxindoles. Org. Biomol. Chem. 2021, 19, 7768–7791. [Google Scholar] [CrossRef]
- Palmisano, G.; Annunziata, R.; Papeo, G.; Sisti, M. Oxindole alkaloids. A novel non-biomimetic entry to (−)-Horsfiline. Tetrahedron Asymmetry 1996, 7, 1–4. [Google Scholar] [CrossRef]
- Siitonen, J.; Lira, S.; Yousufuddin, M.; Kürti, L. Total synthesis of isatindigotindoline C. Org. Biomol. Chem. 2020, 18, 2051–2053. [Google Scholar] [CrossRef]
- Yu, Z.; Liu, X.; Pan, B.; Chen, B.; Zhou, Y.; Wang, H. Construction of pyrrolidinyl spirooxindoles via a 1,3-dipolar cycloaddition reaction of (E)-N-Boc-3-alkylidene-indolin-2(3H)ones with azomethine ylides. Synth. Commun. 2014, 44, 530–539. [Google Scholar] [CrossRef]
- Huang, Y.; Fang, H.; Huang, Y.; Sun, J.; Yan, C. Synthesis of 7′-Arylidenespiro[indoline-3,1′-pyrrolizines] and 7′-Arylidenespiro[indene-2,1′-pyrrolizines] via [3 + 2] Cycloaddition and β-C–H Functionalized Pyrrolidine. J. Org. Chem. 2019, 84, 12437–12451. [Google Scholar] [CrossRef]
- Day, J.; Uroos, M.; Castledine, R.A.; Lewis, W.; McKeever-Abbas, B.; Dowden, J. Alkaloid inspired spirocyclic oxindoles from 1,3-dipolar cycloaddition of pyridinium ylides. Org. Biomol. Chem. 2013, 11, 6502–6509. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhang, Y.; Shen, G.; Yan, C. Molecular Diversity of 1,3-Dipolar Cycloaddition of Quinolinium Ylides with Isatylidene Malononitriles. ChemistrySelect 2017, 2, 10835–10839. [Google Scholar] [CrossRef]
- Wu, L.; Sun, J.; Yan, C. Facile synthesis of spiro[indoline-3,3′-pyrrolo [1,2-a]quinolines] and spiro[indoline-3,1′-pyrrolo [2,1-a]isoquinolines] via 1,3-dipolar cycloaddition reactions of heteroaromatic ammonium salts with 3-phenacylideneoxindoles Org. Biomol. Chem. 2012, 10, 9452–9463. [Google Scholar] [CrossRef]
- Novotortsev, V.K.; Kukushkin, M.E.; Tafeenko, V.A.; Skvortsov, D.A.; Kalinina, M.A.; Timoshenko, R.V.; Chmelyuk, N.S.; Vasilyeva, L.A.; Tarasevich, B.N.; Gorelkin, P.V.; et al. Dispirooxindoles Based on 2-Selenoxo-Imidazolidin-4-Ones: Synthesis, Cytotoxicity and ROS Generation Ability Int. J. Mol. Sci. 2021, 22, 2613. [Google Scholar] [CrossRef]
- Ivanenkov, Y.A.; Vasilevski, S.V.; Beloglazkina, E.K.; Kukushkin, M.E.; Machulkin, A.E.; Veselov, M.S.; Chufarova, N.V.; Chernyagin, E.S.; Vanzcool, A.S.; Zyk, N.V.; et al. Design, synthesis and biological evaluation of novel potent MDM2/p53 small-molecule inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 404–409. [Google Scholar] [CrossRef]
- Mathusalini, S.; Arasakumar, T.; Lakshmi, K.; Lin, C.-H.; Mohan, P.S.; Ramnath, M.G.; Thirugnanasampandan, R. Synthesis and biological evaluation of new spirooxindoles with embedded pharmacophores. New J. Chem. 2016, 40, 5164–5169. [Google Scholar] [CrossRef]
- Teja, C.; Babu, S.N.; Noor, A.; Daniel, J.A.; Devi, S.A.; Nawaz Khan, F.R. Cu/TEMPO catalyzed dehydrogenative 1,3-dipolar cycloaddition in the synthesis of spirooxindoles as potential antidiabetic agents. RSC Adv. 2020, 10, 12262–12271. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Huang, Y.-X.; Suna, J.; Yan, C.-G. Diastereoselective synthesis of dispirooxindoles via [3+2] cycloaddition of azomethine ylides to 3-phenacylideneoxindoles and evaluation of their cytotoxicity. RSC Adv. 2018, 8, 23990–23995. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.; Zhao, Q.; Yu, M.; Guo, L.; Chang, H.; Jiang, X.; Luo, Y.; Huang, W.; He, G. Design and synthesis of novel spirooxindole–indenoquinoxaline derivatives as novel tryptophanyl-tRNA synthetase inhibitors. Mol. Divers. 2020, 24, 1043–1063. [Google Scholar] [CrossRef]
- Raunak; Kumar, V.; Mukherjee, S.; Poonam; Prasad, A.; Olsen, C.; Schäffer, S.; Sharma, S.; Watterson, A.; Errington, W.; et al. Microwave mediated synthesis of spiro-(indoline-isoxazolidines): Mechanistic study and biological activity evaluation. Tetrahedron 2005, 61, 5687–5697. [Google Scholar] [CrossRef]
- Malhotra, S.; Balwani, S.; Dhawan, A.; Raunak; Kumar, Y.; Singh, B.K.; Olsen, C.E.; Prasad, A.K.; Parmar, V.S.; Ghosh, B. Design, synthesis and biological activity evaluation of regioisomeric spiro-(indoline-isoxazolidines) in the inhibition of TNF-α-induced ICAM-1 expression on human endothelial cells. MedChemComm 2012, 3, 1536–1547. [Google Scholar] [CrossRef]
- Gupta, E.; Nair, S.; Kant, R.; Mohanan, K. Additive-Free Three-Component Synthesis of Spiro-isoxazolidine-oxindoles Employing Trifluorodiazoethane. J. Org. Chem. 2018, 83, 14811–14819. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Inada, M.; Okamoto, I.; Morita, N.; Tamura, O. Synthesis of Maremycins A and D1 via Cycloaddition of a Nitrone with (E)-3-Ethylidene-1-methylindolin-2-one. Org. Lett. 2008, 10, 2043–2046. [Google Scholar] [CrossRef]
- Zhang, D.; Yin, C.; Zhou, Y.; Xu, Y.; Lin, L.; Liu, X.; Feng, X. Chiral N,N′-dioxide/Co(II)-promoted asymmetric 1,3-dipolar cycloaddition of nitrones with methyleneindolinones. Chem. Commun. 2017, 53, 7925–7928. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, A.; Mao, H.; Mao, Z.; Li, W.; Hu, H.; Zhu, C.; Cheng, Y. Enantioselective Construction of Spirooxindole Derivatives through [3+2] Annulation Catalyzed by a Bisthiourea as a Multiple-Hydrogen-Bond Donor. Chem. Eur. J. 2013, 19, 1914–1918. [Google Scholar] [CrossRef]
- Ueda, T.; Inada, M.; Morita, N.; Tamura, O. Total Synthesis of Maremycins A and D1 Using Chiral and Cyclic Nitrone with (E)-3-Ethylidene-1-methylindolin-2-one. Heterocycles 2015, 90, 1179–1195. [Google Scholar] [CrossRef]
- Malinina, J.; Tran, T.Q.; Stepakov, A.V.; Gurzhiy, V.V.; Starova, G.L.; Kostikov, R.R.; Molchanov, A.P. [3+2] Cycloaddition reactions of arylallenes with C-(N-arylcarbamoyl)- and C,C-bis(methoxycarbonyl)nitrones and subsequent rearrangements. Tetrahedron Lett. 2014, 55, 3663–3666. [Google Scholar] [CrossRef]
- Efremova, M.M.; Novikov, A.S.; Kostikov, R.R.; Panikorovsky, T.L.; Ivanov, A.V.; Molchanov, A.P. Regio- and diastereoselectivity of the cycloaddition of nitrones with N-propadienylindole and pyrroles. Tetrahedron 2018, 74, 174–183. [Google Scholar] [CrossRef]
- Molchanov, A.P.; Malinina, Y.V.; Kostikov, R.R.; Stepakov, A.V. Regioselective cycloaddition of C,N-diarylnitrones to arylallenes and of N-aryl-C-carbamoylnitrones to methyl buta-2,3-dienoate. Russ. J. Org. Chem. 2015, 51, 368–372. [Google Scholar] [CrossRef]
- Berthet, M.; Cheviet, T.; Dujardin, G.; Parrot, I.; Martinez, J. Isoxazolidine: A Privileged Scaffold for Organic and Medicinal Chemistry. Chem. Rev. 2016, 116, 15235–15283. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Giofre, S.V.; Romeo, R.; Romeo, G.; Chiacchio, U. Isoxazolidines as Biologically Active Compounds. Curr. Org. Synth. 2016, 13, 726–749. [Google Scholar] [CrossRef] [Green Version]
- Romeo, R.; Giofrè, S.V.; Carnovale, C.; Campisi, A.; Parenti, R.; Bandini, R.; Chiacchio, M.A. Synthesis and biological evaluation of 3-hydroxymethyl-5-(1H-1,2,3-triazol) isoxazolidines. Bioorg. Med. Chem. 2013, 21, 7929–7937. [Google Scholar] [CrossRef]
- Grabkowska-Dru’zyc, M.; Andrei, G.; Schols, D.; Snoeck, R.; Piotrowska, D.G. Isoxazolidine Conjugates of N3-Substituted 6-Bromoquinazolinones—Synthesis, Anti-Varizella-Zoster Virus, and Anti-Cytomegalovirus Activity. Molecules 2018, 23, 1889. [Google Scholar] [CrossRef] [Green Version]
- Giofrè, S.V.; Cirmi, S.; Mancuso, R.; Nicolò, F.; Lanza, G.; Legnani, L.; Campisi, A.; Chiacchio, M.A.; Navarra, M.; Gabriele, B.; et al. Synthesis of spiro[isoindole-1,5′-isoxazolidin]-3(2H)-ones as potential inhibitors of the MDM2-p53 interaction. Beilstein J. Org. Chem. 2016, 12, 2793–2807. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Song, S.; Shu, S.; Miao, Z.; Zhang, A.; Ding, C. Novel spirobicyclic artemisinin analogues (artemalogues): Synthesis and antitumor activities. Eur. J. Med. Chem. 2015, 103, 17–28. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Andrei, G.; Schols, D.; Snoeck, R.; Łysakowska, M. Synthesis, anti-varicella-zoster virus and anti-cytomegalovirus activity of quinazoline-2,4-diones containing isoxazolidine and phosphonate substructures. Eur. J. Med. Chem. 2017, 126, 84–100. [Google Scholar] [CrossRef]
- Sirotkina, E.V.; Efremova, M.M.; Novikov, A.S.; Zarubaev, V.V.; Orshanskaya, I.R.; Starova, G.L.; Kostikov, R.R.; Molchanov, A.P. Regio- and diastereoselectivity of the cycloaddition of aldonitrones with benzylidenecyclopropane: An experimental and theoretical study. Tetrahedron 2017, 73, 3025–3030. [Google Scholar] [CrossRef]
- Dmitriev, V.A.; Efremova, M.M.; Novikov, A.S.; Zarubaev, V.V.; Slita, A.V.; Galochkina, A.V.; Starova, G.L.; Ivanov, A.V.; Molchanov, A.P. Highly efficient and stereoselective cycloaddition of nitrones to indolyl- and pyrrolylacrylates. Tetrahedron Lett. 2018, 59, 2327–2331. [Google Scholar] [CrossRef]
- Molchanov, A.P.; Lukina, V.M.; Efremova, M.M.; Muryleva, A.A.; Slita, A.V.; Zarubaev, V.V. The 1,3-dipolar cycloaddition of adamantine-derived nitrones with maleimides. Synth. Commun. 2020, 50, 1367–1374. [Google Scholar] [CrossRef]
- Efremova, M.M.; Molchanov, A.P.; Novikov, A.S.; Starova, G.L.; Muryleva, A.A.; Slita, A.V.; Zarubaev, V.V. 1,3-Dipolar cycloaddition of N-allyl substituted polycyclic derivatives of isoindole-1,3-dione with nitrones and nitrile oxides: An experimental and theoretical investigation. Tetrahedron 2020, 76, 131104. [Google Scholar] [CrossRef]
- Singh, G.; Singh, G.; Bhatti, R.; Gupta, V.; Mahajan, A.; Singh, P.; Singh Ishar, M.P. Rationally designed benzopyran fused isoxazolidines and derived β2,3,3-amino alcohols as potent analgesics: Synthesis, biological evaluation and molecular docking analysis. Eur. J. Med. Chem. 2017, 127, 210–222. [Google Scholar] [CrossRef]
- Shibue, T.; Hirai, T.; Okamoto, I.; Morita, N.; Masu, H.; Azumaya, I.; Tamura, O. Total Syntheses of Tubulysins. Chem. Eur. J. 2010, 16, 11678–11688. [Google Scholar] [CrossRef]
- Evans, D.A.; Song, H.-J.; Fandrick, K.R. Enantioselective Nitrone Cycloadditions of α,β-Unsaturated 2-Acyl Imidazoles Catalyzed by Bis(oxazolinyl)pyridine−Cerium(IV) Triflate Complexes. Org. Lett. 2006, 8, 3351–3354. [Google Scholar] [CrossRef]
- Diethelm, S.; Carreira, E.M. Total Synthesis of Gelsemoxonine through a Spirocyclopropane Isoxazolidine Ring Contraction. J. Am. Chem. Soc. 2015, 137, 6084–6096. [Google Scholar] [CrossRef]
- Diethelm, S.; Schoenebeck, F.; Carreira, E.M. Mechanistic Insight into the Spirocyclopropane Isoxazolidine Ring Contraction. Org. Lett. 2014, 16, 960–963. [Google Scholar] [CrossRef]
- Romeo, G.; Iannazzo, D.; Piperno, A.; Romeo, R.; Corsaro, A.; Rescifina, A.; Chiacchio, U. C-Alkoxycarbonyl Nitrones: Building Blocks for the Synthesis of Butenolides, Lactams and Modified Nucleosides. Mini-Rev. Org. Chem. 2005, 2, 59–77. [Google Scholar] [CrossRef]
- Padwa, A.; Koehler, K.F.; Rodrigues, A. Nitrone cycloaddition. New approach to .beta.-lactams. J. Am. Chem. Soc. 1981, 103, 4974–4975. [Google Scholar] [CrossRef]
- Cordero, F.M.; Pisaneschi, F.; Goti, A.; Ollivier, J.; Salaün, J.; Brandi, A. New synthesis of β-lactams by ethylene extrusion from spirocyclopropane isoxazolidines. J. Am. Chem. Soc. 2000, 122, 8075–8076. [Google Scholar] [CrossRef]
- Yadav, S.; Taylor, C.M. Synthesis of Orthogonally Protected (2S)-2-Amino-adipic Acid (α-AAA) and (2S,4R)-2-Amino-4-hydroxyadipic Acid (Ahad). J. Org. Chem. 2013, 78, 5401–5409. [Google Scholar] [CrossRef]
- Teterina, P.S.; Efremova, M.M.; Sirotkina, E.V.; Novikov, A.S.; Khoroshilova, O.V.; Molchanov, A.P. A highly efficient and stereoselective cycloaddition of nitrones to N-arylitaconimides. Tetrahedron Lett. 2019, 60, 151063. [Google Scholar] [CrossRef]
- Sirotkina, E.V.; Efremova, M.M.; Starova, G.L.; Kuznetsov, M.A.; Molchanov, A.P. Cycloaddition of nitrones to 1,3-diarylpropenones and subsequent transformations of the resulting isoxazolidines. Chem. Het. Comp. 2020, 56, 1193–1201. [Google Scholar] [CrossRef]
- Burdisso, M.; Gamba, A.; Gandolf, R.; Pevarello, P. Stereospecificity of 1,3-dipolar cycloadditions of cyclic nitrones to (E) and (Z)-β-nitrostyrenes. Tetrahedron 1987, 43, 1835–1846. [Google Scholar] [CrossRef]
- Yuan, W.; Cui, T.; Liu, W.; Wen, L.; Li, M. When Ethyl Isocyanoacetate Meets Isatins: A 1,3-Dipolar/Inverse 1,3-Dipolar/Olefination Reaction for Access to 3-Ylideneoxindoles. Org. Lett. 2018, 20, 1513–1516. [Google Scholar] [CrossRef]
- Song, Z.; Chen, C.; Liu, J.; Wen, X.; Sun, H.; Yuan, H. Design, synthesis, and biological evaluation of (2E)-(2-oxo-1, 2-dihydro-3H-indol-3-ylidene)acetate derivatives as anti-proliferative agents through ROS-induced cell apoptosis. Eur. J. Med. Chem. 2016, 124, 809–819. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Conditions | Ratio 3a:3′a:4a 1 | Isolated Yield, % | ||
|---|---|---|---|---|---|
| 3a | 3′a | 4a | |||
| 1 | CH3CN, 82 °C, 14 h | - 2 | 67 | - | - |
| 2 | PhMe, 80 °C, 14 h | 4:1:0.3 | 70 | 18 | |
| 3 | PhMe, 110 °C, 7 h | - 2 | 45 | - | - |
| 4 | CH2Cl2, rt, 7 h | 1:4:0 | 11 | 73 | - |

| Entry | Dipolarophile | R | Nitrone | R’ | Products | Ratio 3a:3′a:4a 1 | Isolated Yield, % | |
|---|---|---|---|---|---|---|---|---|
| 3 | (3′ + 4) | |||||||
| 1 | 1a | H | 2a | H | 3a, 3′a, 4a | 4:1:0.3 | 70 | 18 |
| 2 | 1a | H | 2b | Cl | 3b, 3′b, 4b | 5:1:0.2 | 70 | 16 |
| 3 | 1a | H | 2c | Me | 3c, 3′c, 4c | 5:1:0.5 | 57 | 24 2 |
| 4 | 1a | H | 2d | MeO | 3d, 3′d, 4d | 4:1:0.3 | 69 | 19 |
| 5 | 1b | Me | 2a | H | 3e, 3′e, 4e | 5:1:0.2 | 74 | 13 |
| 6 | 1b | Me | 2b | Cl | 3f, 3′f, 4f | 5:1:0.2 | 65 | 17 |
| 7 | 1b | Me | 2c | Me | 3g, 3′g, 4g | 3.5:1:0.3 | 55 | 20 |
| 8 | 1b | Me | 2d | MeO | 3h, 3′h, 4h | 3.2:1:0.2 | 58 | 12 3 |

| Entry | Dipolarophile | R | Nitrone | R’ | Products | Ratio 3a:3′a 1 | Isolated Yield, % | |
|---|---|---|---|---|---|---|---|---|
| 3 | 3′ | |||||||
| 1 | 1a | H | 2a | H | 3a, 3′a | 1:4 | 11 | 73 |
| 2 | 1a | H | 2b | Cl | 3b, 3′b | 1:4 | 17 | 58 |
| 3 | 1b | Me | 2b | Cl | 3f, 3′f | 1:4 | 14 | 61 |

| Entry | Dipolarophile | R | Nitrone | R’ | Product | Isolated Yield of 6, % |
|---|---|---|---|---|---|---|
| 1 | 1a | H | 5a | H | 6a | 95 |
| 2 | 1a | H | 5b | Me | 6b | 66 |
| 3 | 1a | H | 5c | MeO | 6c | 72 |
| 4 | 1b | Me | 5a | H | 6d | 92 |
| 5 | 1b | Me | 5b | Me | 6e | 77 |
| 6 | 1b | Me | 5c | MeO | 6f | 82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karcev, D.D.; Efremova, M.M.; Molchanov, A.P.; Rostovskii, N.V.; Kryukova, M.A.; Bunev, A.S.; Khochenkov, D.A. Selective and Reversible 1,3-Dipolar Cycloaddition of 2-(2-Oxoindoline-3-ylidene)acetates with Nitrones in the Synthesis of Functionalized Spiroisoxazolidines. Int. J. Mol. Sci. 2022, 23, 12639. https://doi.org/10.3390/ijms232012639
Karcev DD, Efremova MM, Molchanov AP, Rostovskii NV, Kryukova MA, Bunev AS, Khochenkov DA. Selective and Reversible 1,3-Dipolar Cycloaddition of 2-(2-Oxoindoline-3-ylidene)acetates with Nitrones in the Synthesis of Functionalized Spiroisoxazolidines. International Journal of Molecular Sciences. 2022; 23(20):12639. https://doi.org/10.3390/ijms232012639
Chicago/Turabian StyleKarcev, Dmitriy D., Mariia M. Efremova, Alexander P. Molchanov, Nikolai V. Rostovskii, Mariya A. Kryukova, Alexander S. Bunev, and Dmitry A. Khochenkov. 2022. "Selective and Reversible 1,3-Dipolar Cycloaddition of 2-(2-Oxoindoline-3-ylidene)acetates with Nitrones in the Synthesis of Functionalized Spiroisoxazolidines" International Journal of Molecular Sciences 23, no. 20: 12639. https://doi.org/10.3390/ijms232012639