
Mutation Hotspot for Changing the Substrate Specificity of β-N-Acetylhexosaminidase: A Library of GlcNAcases
 , , , , and
, , , , and 
Abstract
:1. Introduction
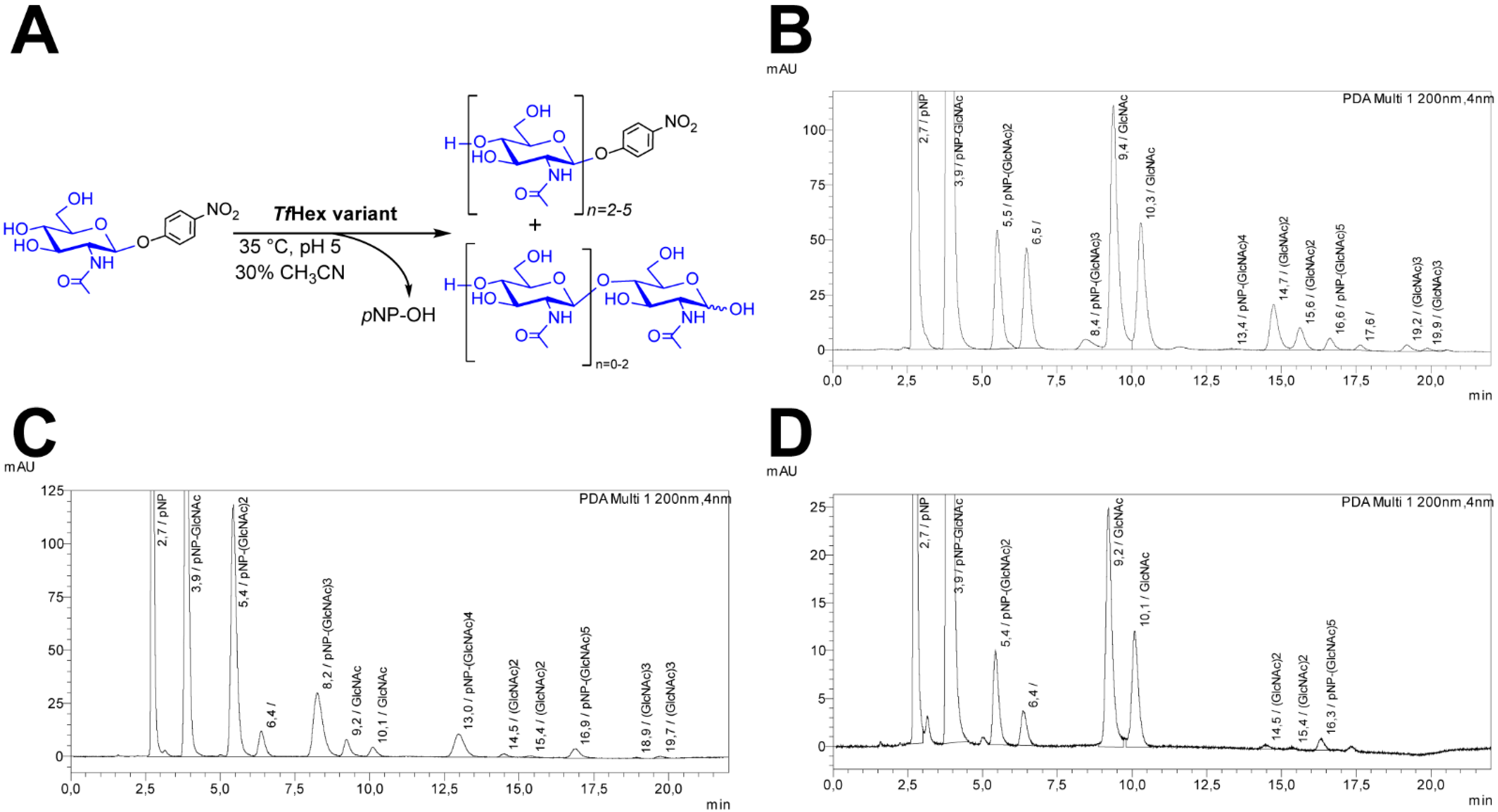
2. Results
2.1. Site-Saturation Mutagenesis Approach—A Dead-End Street?
2.2. Site-Directed Mutagenesis Approach
2.2.1. In Silico Rational Design of Prospective TfHex Mutants
2.2.2. Preparation of TfHex Mutants by Site-Directed Mutagenesis
2.2.3. Production and Characterization of Site-Directed Mutants of TfHex
2.2.4. Kinetic Analysis and Transglycosylation Potential of the Best Mutants
3. Discussion
4. Materials and Methods
4.1. General
4.2. Preparation and Cloning of Mutant Genes
4.3. Transformation of P. pastoris Host with Mutant Genes
4.4. Deep-Well Screening for Heterologous Expression of β-N-Acetylhexosaminidase Variants in P. pastoris
4.5. Heterologous Expression of β-N-Acetylhexosaminidase Variants in P. pastoris, Screening and Purification
4.6. Isolation of Chromosomal DNA
4.7. Amplification and Ligation of Mutant Genes into the Plasmid for Sequencing
4.8. Protein Characterization
4.9. Enzyme Activity Assay
4.10. Analytical Transglycosylation Reactions
4.11. Molecular Modeling, Docking, and Molecular Dynamics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muschiol, J.; Vuillemin, M.; Meyer, A.S.; Zeuner, B. β-N-Acetylhexosaminidases for carbohydrate synthesis via trans-glycosylation. Catalysts 2020, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Slámová, K.; Bojarová, P.; Petrásková, L.; Křen, V. β-N-Acetylhexosaminidase: What’s in a name…? Biotechnol. Adv. 2010, 28, 682–693. [Google Scholar] [CrossRef]
- Rauvolfová, J.; Kuzma, M.; Weignerová, L.; Fialová, P.; Přikrylová, V.; Pišvejcová, A.; Macková, M.; Křen, V. β-N-Acetylhexosaminidase-catalysed synthesis of non-reducing oligosaccharides. J. Mol. Catal. B Enzym. 2004, 29, 233–239. [Google Scholar] [CrossRef]
- Fialová, P.; Weignerová, L.; Rauvolfová, J.; Přikrylová, V.; Pišvejcová, A.; Ettrich, R.; Kuzma, M.; Sedmera, P.; Křen, V. Hydrolytic and transglycosylation reactions of N-acyl modified substrates catalysed by β-N-acetylhexosaminidases. Tetrahedron 2004, 60, 693–701. [Google Scholar] [CrossRef]
- Slámová, K.; Bojarová, P. Engineered N-acetylhexosamine-active enzymes in glycoscience. BBA—Gen. Subj. 2017, 1861, 2070–2087. [Google Scholar] [CrossRef]
- Bojarová, P.; Bruthans, J.; Křen, V. β-N-Acetylhexosaminidases—the wizards of glycosylation. Appl. Microbiol. Biotechnol. 2019, 103, 7869–7881. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jin, L.; Jiang, X.; Guo, L.; Gu, G.; Xu, L.; Lu, L.; Wang, F.; Xiao, M. Converting a β-N-acetylhexosaminidase into two trans-β-N-acetylhexosaminidases by domain-targeted mutagenesis. Appl. Microbiol. Biotechnol. 2020, 104, 661–673. [Google Scholar] [CrossRef]
- Jamek, S.B.; Muschiol, J.; Holck, J.; Zeuner, B.; Busk, P.K.; Mikkelsen, J.D.; Meyer, A.S. Loop protein engineering for improved transglycosylation activity of a b-N-acetylhexosaminidase. ChemBioChem 2018, 19, 1858–1865. [Google Scholar] [CrossRef]
- Weignerová, L.; Vavrušková, P.; Pišvejcová, A.; Thiem, J.; Křen, V. Fungal β-N-acetylhexosaminidases with high β-N-acetylgalactosaminidase activity and their use for synthesis of β-GalNAc-containing oligosaccharides. Carbohydr. Res. 2003, 338, 1003–1008. [Google Scholar] [CrossRef]
- Bojarová, P.; Kulik, N.; Slámová, K.; Hubálek, M.; Kotik, M.; Cvačka, J.; Pelantová, H.; Křen, V. Selective β-N-acetylhexosaminidase from Aspergillus versicolor—A tool for producing bioactive carbohydrates. Appl. Microbiol. Biotechnol. 2019, 103, 1737–1753. [Google Scholar] [CrossRef]
- Nekvasilová, P.; Andreasová, I.; Petrásková, L.; Pelantová, H.; Křen, V.; Bojarová, P. A novel enzymatic tool for transferring GalNAc moiety onto challenging acceptors. BBA—Proteins Proteom. 2020, 1868, 140319. [Google Scholar] [CrossRef]
- Chen, X.; Xu, L.; Jin, L.; Sun, B.; Gu, G.; Lu, L.; Xiao, M. Efficient and regioselective synthesis of β-GalNAc/GlcNAc-lactose by a bifunctional transglycosylating β-N-acetylhexosaminidase from Bifidobacterium bifidum. Appl. Environ. Microbiol. 2016, 82, 5642. [Google Scholar] [CrossRef] [Green Version]
- Kurakake, M.; Goto, T.; Ashiki, K.; Suenaga, Y.; Komaki, T. Synthesis of new glycosides by transglycosylation of N-acetylhexosaminidase from Serratia marcescens YS-1. J. Agric. Food Chem. 2003, 51, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Visnapuu, T.; Teze, D.; Kjeldsen, C.; Lie, A.; Duus, J.Ø.; André-Miral, C.; Pedersen, L.H.; Stougaard, P.; Svensson, B. Identification and characterization of a β-N-acetylhexosaminidase with a biosynthetic activity from the marine bacterium Paraglaciecola hydrolytica S66T. Int. J. Mol. Sci. 2020, 21, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, C.; Petricevic, M.; John, A.; Goddard-Borger, E.D.; Davies, G.J.; Williams, S.J. Structural and mechanistic insights into a Bacteroides vulgatus retaining N-acetyl-β-galactosaminidase that uses neighbouring group participation. Chem. Commun. 2016, 52, 11096–11099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noach, I.; Pluvinage, B.; Laurie, C.; Abe, K.T.; Alteen, M.G.; Vocadlo, D.J.; Boraston, A.B. The Details of glycolipid glycan hydrolysis by the structural analysis of a family 123 glycoside hydrolase from Clostridium perfringens. J. Mol. Biol. 2016, 428, 3253–3265. [Google Scholar] [CrossRef] [Green Version]
- Sumida, T.; Fujimoto, K.; Ito, M. Molecular cloning and catalytic mechanism of a novel glycosphingolipid-degrading β-N-acetylgalactosaminidase from Paenibacillus sp. TS12. J. Biol. Chem. 2011, 286, 14065–14072. [Google Scholar] [CrossRef] [Green Version]
- Gloster, T.M.; Vocadlo, D.J. Mechanism, structure, and inhibition of O-GlcNAc processing enzymes. Curr. Signal Transduct. Ther. 2010, 5, 74–91. [Google Scholar] [CrossRef] [Green Version]
- Krejzová, J.; Šimon, P.; Kalachova, L.; Kulik, N.; Bojarová, P.; Marhol, P.; Pelantová, H.; Cvačka, J.; Ettrich, R.; Slámová, K.; et al. Inhibition of GlcNAc-processing glycosidases by C-6-azido-NAG-thiazoline and its derivatives. Molecules 2014, 19, 3471–3488. [Google Scholar] [CrossRef] [Green Version]
- Bojarová, P.; Křenek, K.; Kuzma, M.; Petrásková, L.; Bezouška, K.; Namdjou, D.-J.; Elling, L.; Křen, V. N-Acetylhexosamine triad in one molecule: Chemoenzymatic introduction of 2-acetamido-2-deoxy-β-d-galactopyranosyluronic acid residue into a complex oligosaccharide. J. Mol. Catal. B Enzym. 2008, 50, 69–73. [Google Scholar] [CrossRef]
- Bojarová, P.; Slámová, K.; Křenek, K.; Gažák, R.; Kulik, N.; Ettrich, R.; Pelantová, H.; Kuzma, M.; Riva, S.; Adámek, D.; et al. Charged hexosaminides as new substrates for β-N-acetylhexosaminidase-catalyzed synthesis of immunomodulatory disaccharides. Adv. Synth. Catal. 2011, 353, 2409–2420. [Google Scholar] [CrossRef]
- Slámová, K.; Gažák, R.; Bojarová, P.; Kulik, N.; Ettrich, R.; Pelantová, H.; Sedmera, P.; Křen, V. 4-Deoxy-substrates for β-N-acetylhexosaminidases: How to make use of their loose specificity. Glycobiology 2010, 20, 1002–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojarová, P.; Kulik, N.; Hovorková, M.; Slámová, K.; Pelantová, H.; Křen, V. The β-N-acetylhexosaminidase in the synthesis of bioactive glycans: Protein and reaction engineering. Molecules 2019, 24, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettrich, R.; Kopecký, V.; Hofbauerová, K.; Baumruk, V.; Novák, P.; Pompach, P.; Man, P.; Plíhal, O.; Kutý, M.; Kulik, N.; et al. Structure of the dimeric N-glycosylated form of fungal β-N-acetylhexosaminidase revealed by computer modeling, vibrational spectroscopy, and biochemical studies. BMC Struct. Biol. 2007, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Ryšlavá, H.; Kalendová, A.; Doubnerová, V.; Skočdopol, P.; Kumar, V.; Kukačka, Z.; Pompach, P.; Vaněk, O.; Slámová, K.; Bojarová, P.; et al. Enzymatic characterization and molecular modeling of an evolutionarily interesting fungal β-N-acetylhexosaminidase. FEBS J. 2011, 278, 2469–2484. [Google Scholar] [CrossRef] [PubMed]
- Fialová, P.; Carmona, A.T.; Robina, I.; Ettrich, R.; Sedmera, P.; Přikrylová, V.; Petrásková-Hušáková, L.; Křen, V. Glycosyl azide—A novel substrate for enzymatic transglycosylations. Tetrahedron Lett. 2005, 46, 8715–8718. [Google Scholar] [CrossRef]
- Slámová, K.; Krejzová, J.; Marhol, P.; Kalachova, L.; Kulik, N.; Pelantová, H.; Cvačka, J.; Křen, V. Synthesis of derivatized chitooligomers using transglycosidases engineered from the fungal GH20 β-N-acetylhexosaminidase. Adv. Synth. Catal. 2015, 357, 1941–1950. [Google Scholar] [CrossRef]
- Kapešová, J.; Petrásková, L.; Kulik, N.; Straková, Z.; Bojarová, P.; Markošová, K.; Rebroš, M.; Křen, V.; Slámová, K. Transglycosidase activity of glycosynthase-type mutants of a fungal GH20 β-N-acetylhexosaminidase. Int. J. Biol. Macromol. 2020, 161, 1206–1215. [Google Scholar] [CrossRef]
- Mackenzie, L.F.; Wang, Q.; Warren, R.A.J.; Withers, S.G. Glycosynthases: Mutant glycosidases for oligosaccharide synthesis. J. Am. Chem. Soc. 1998, 120, 5583–5584. [Google Scholar] [CrossRef]
- Malet, C.; Planas, A. From β-glucanase to β-glucansynthase: Glycosyl transfer to α-glycosyl fluorides catalyzed by a mutant endoglucanase lacking its catalytic nucleophile. FEBS Lett. 1998, 440, 208–212. [Google Scholar] [CrossRef]
- Mészáros, Z.; Petrásková, L.; Kulik, N.; Pelantová, H.; Bojarová, P.; Křen, V.; Slámová, K. Hypertransglycosylating variants of the GH20 β-N-acetylhexosaminidase for the synthesis of chitooligomers. Adv. Synth. Catal. 2022, 364, 2009–2022. [Google Scholar] [CrossRef]
- Nekvasilová, P.; Kulik, N.; Rychlá, N.; Pelantová, H.; Petrásková, L.; Bosáková, Z.; Cvačka, J.; Slámová, K.; Křen, V.; Bojarová, P. How site-directed mutagenesis boosted selectivity of a promiscuous enzyme. Adv. Synt. Catal. 2020, 362, 4138–4150. [Google Scholar] [CrossRef]
- Bojarová, P.; Chytil, P.; Mikulová, B.; Bumba, L.; Konefał, R.; Pelantová, H.; Krejzová, J.; Slámová, K.; Petrásková, L.; Kotrchová, L.; et al. Glycan-decorated HPMA copolymers as high-affinity lectin ligands. Polym. Chem. 2017, 8, 2647–2658. [Google Scholar] [CrossRef] [Green Version]
- Bojarová, P.; Tavares, M.R.; Laaf, D.; Bumba, L.; Petrásková, L.; Konefał, R.; Bláhová, M.; Pelantová, H.; Elling, L.; Etrych, T.; et al. Biocompatible glyconanomaterials based on HPMA-copolymer for specific targeting of galectin-3. J. Nanobiotechnol. 2018, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Laaf, D.; Bojarová, P.; Mikulová, B.; Pelantová, H.; Křen, V.; Elling, L. Two-step enzymatic synthesis of β-d-N-acetylgalactosamine-(1 → 4)-d-N-acetylglucosamine (LacdiNAc) chitooligomers for deciphering galectin binding behavior. Adv. Synth. Catal. 2017, 359, 2101–2108. [Google Scholar] [CrossRef] [Green Version]
- Drozdová, A.; Bojarová, P.; Křenek, K.; Weignerová, L.; Henßen, B.; Elling, L.; Christensen, H.; Jensen, H.H.; Pelantová, H.; Kuzma, M.; et al. Enzymatic synthesis of dimeric glycomimetic ligands of NK cell activation receptors. Carbohydr. Res. 2011, 346, 1599–1609. [Google Scholar] [CrossRef]
- Kulik, N.; Slámová, K.; Ettrich, R.; Křen, V. Computational study of β-N-acetylhexosaminidase from Talaromyces flavus, a glycosidase with high substrate flexibility. BMC Bioinf. 2015, 16, 28. [Google Scholar] [CrossRef] [Green Version]
- Škerlová, J.; Bláha, J.; Pachl, P.; Hofbauerová, K.; Kukačka, Z.; Man, P.; Pompach, P.; Novák, P.; Otwinowski, Z.; Brynda, J.; et al. Crystal structure of native b-N-acetylhexosaminidase isolated from Aspergillus oryzae sheds light onto its substrate specificity, high stability, and regulation by propeptide. FEBS J. 2018, 285, 580–598. [Google Scholar] [CrossRef] [Green Version]
- Slámová, K.; Bojarová, P.; Gerstorferová, D.; Fliedrová, B.; Hofmeisterová, J.; Fiala, M.; Pompach, P.; Křen, V. Sequencing, cloning and high-yield expression of a fungal β-N-acetylhexosaminidase in Pichia pastoris. Protein Expression Purif. 2012, 82, 212–217. [Google Scholar] [CrossRef]
- Karbalaei, M.; Rezaee, S.A.; Farsiani, H. Pichia pastoris: A highly successful expression system for optimal synthesis of heterologous proteins. J. Cell. Physiol. 2020, 235, 5867–5881. [Google Scholar] [CrossRef]
- Marx, H.; Mecklenbräuker, A.; Gasser, B.; Sauer, M.; Mattanovich, D. Directed gene copy number amplification in Pichia pastoris by vector integration into the ribosomal DNA locus. FEMS Yeast Res. 2009, 9, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reetz, M.T. Laboratory evolution of stereoselective enzymes as a means to expand the toolbox of organic chemists. Tetrahedron 2012, 68, 7530–7548. [Google Scholar] [CrossRef]
- Reetz, M.T.; Kahakeaw, D.; Lohmer, R. Addressing the numbers problem in directed evolution. ChemBioChem 2008, 9, 1797–1804. [Google Scholar] [CrossRef]
- Weis, R.; Luiten, R.; Skranc, W.; Schwab, H.; Wubbolts, M.; Glieder, A. Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res. 2004, 5, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansur, M.; Cabello, C.; Hernández, L.; País, J.; Varas, L.; Valdés, J.; Terrero, Y.; Hidalgo, A.; Plana, L.; Besada, V.; et al. Multiple gene copy number enhances insulin precursor secretion in the yeast Pichia pastoris. Biotechnol. Lett. 2005, 27, 339–345. [Google Scholar] [CrossRef]
- Vassileva, A.; Arora Chugh, D.; Swaminathan, S.; Khanna, N. Effect of copy number on the expression levels of hepatitis B surface antigen in the methylotrophic yeast Pichia pastoris. Protein Expression Purif. 2001, 21, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, B.; Pei, H.; Lv, J.; Yang, W.; Cao, Y.; Dong, B. Directed evolution of Penicillium janczewskii zalesk α-galactosidase toward enhanced activity and expression in Pichia pastoris. Appl. Biochem. Biotechnol 2012, 168, 638–650. [Google Scholar] [CrossRef]
- Kao, M.R.; Yu, S.M.; Ho, T.U.D. Improvements of the productivity and saccharification efficiency of the cellulolytic β-glucosidase D2-BGL in Pichia pastoris via directed evolution. Biotechnol. Biofuels 2021, 14, 126. [Google Scholar] [CrossRef]
- Beard, H.; Cholleti, A.; Pearlman, D.; Sherman, W.; Loving, K.A. Applying physics-based scoring to calculate free energies of binding for single amino acid mutations in protein-protein complexes. PLoS ONE 2013, 8, e82849. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhou, Y.; Chen, L.; Chen, W.; Liu, L.; Shen, X.; Zhang, W.; Zhang, J.; Yang, Q. Structural Insights into cellulolytic and chitinolytic enzymes revealing crucial residues of insect β-N-acetyl-D-hexosaminidase. PLoS ONE 2012, 7, e52225. [Google Scholar] [CrossRef]
- Hovorková, M.; Kulik, N.; Konvalinková, D.; Petrásková, L.; Křen, V.; Bojarová, P. Mutagenesis of catalytic nucleophile of β-galactosidase retains residual hydrolytic activity and affords a transgalactosidase. ChemCatChem 2021, 13, 4532–4542. [Google Scholar] [CrossRef]
- Ochoa, R.; Soler, M.A.; Laio, A.; Cossio, P. Assessing the capability of in silico mutation protocols for predicting the finite temperature conformation of amino acids. Phys. Chem. Chem. Phys. 2018, 20, 25901–25909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Garcia-Oliva, C.; Hoyos, P.; Petrásková, L.; Kulik, N.; Pelantová, H.; Cabanillas, A.H.; Rumbero, Á.; Křen, V.; Hernáiz, M.J.; Bojarová, P. Acceptor specificity of β-N-acetylhexosaminidase from Talaromyces flavus: A rational explanation. Int. J. Mol. Sci. 2019, 20, 6181. [Google Scholar] [CrossRef] [Green Version]
- Konagurthu, A.S.; Whisstock, J.C.; Stuckey, P.J.; Lesk, A.M. MUSTANG: A multiple structural alignment algorithm. Proteins 2006, 64, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins: Struct. Funct. Bioinf. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-4: Build 12; S. LLC: New York, NY, USA, 2018.
- Canutescu, A.A.; Shelenkov, A.A.; Dunbrack, R.L., Jr. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003, 12, 2001–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. 9), 114–122. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. In Protein Engineering: Methods and Protocols; Bornscheuer, U.T., Höhne, M., Eds.; Springer: New York, NY, USA, 2018; pp. 43–67. [Google Scholar]
- Turner, P.J. XMGRACE, Version 5.1.19.; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Glu332 Substitution | Yield a [mg] | GlcNAcase [U/mg] | GalNAcase [U/mg] | GalNAcase/ GlcNAcase |
|---|---|---|---|---|
| WT b | 5.1 ± 0.2 | 35 ± 2 | 42 ± 2 | 1.2 |
| Ala | 0.33 ± 0.05 | 54 ± 1 | 12.9 ± 0.5 | 0.24 |
| Asn | 15.5 ± 0.6 | 44 ± 2 | 17.1 ± 0.3 | 0.39 |
| Gln | 9.5 ± 0.3 | 47 ± 1 | 13.6 ± 0.4 | 0.29 |
| Gly | 18.3 ± 0.7 | 68 ± 2 | 17.7 ± 0.6 | 0.26 |
| His | 5.7 ± 0.2 | 8.8 ± 0.1 | 1.32 ± 0.02 | 0.15 |
| Lys | 9.9 ± 0.2 | 1.14 ± 0.04 | 0.21 ± 0.05 | 0.18 |
| Phe | 10.6 ± 0.4 | 7.3 ± 0.1 | 7.4 ± 0.2 | 1.0 |
| Thr | 13.1 ± 0.3 | 138 ± 3 | 30 ± 1 | 0.22 |
| Trp | 13.7 ± 0.3 | 10.5 ± 0.3 | 4.9 ± 0.1 | 0.47 |
| Tyr | 8.8 ± 0.1 | 4.75 ± 0.04 | 6.65 ± 0.05 | 1.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nekvasilová, P.; Kulik, N.; Kotik, M.; Petrásková, L.; Slámová, K.; Křen, V.; Bojarová, P. Mutation Hotspot for Changing the Substrate Specificity of β-N-Acetylhexosaminidase: A Library of GlcNAcases. Int. J. Mol. Sci. 2022, 23, 12456. https://doi.org/10.3390/ijms232012456
Nekvasilová P, Kulik N, Kotik M, Petrásková L, Slámová K, Křen V, Bojarová P. Mutation Hotspot for Changing the Substrate Specificity of β-N-Acetylhexosaminidase: A Library of GlcNAcases. International Journal of Molecular Sciences. 2022; 23(20):12456. https://doi.org/10.3390/ijms232012456
Chicago/Turabian StyleNekvasilová, Pavlína, Natalia Kulik, Michael Kotik, Lucie Petrásková, Kristýna Slámová, Vladimír Křen, and Pavla Bojarová. 2022. "Mutation Hotspot for Changing the Substrate Specificity of β-N-Acetylhexosaminidase: A Library of GlcNAcases" International Journal of Molecular Sciences 23, no. 20: 12456. https://doi.org/10.3390/ijms232012456