
Description of the First Registered Case of Lopes–Maciel–Rodan Syndrome in Russia

 , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Early Disease Manifestation and Clinical History of the Proband
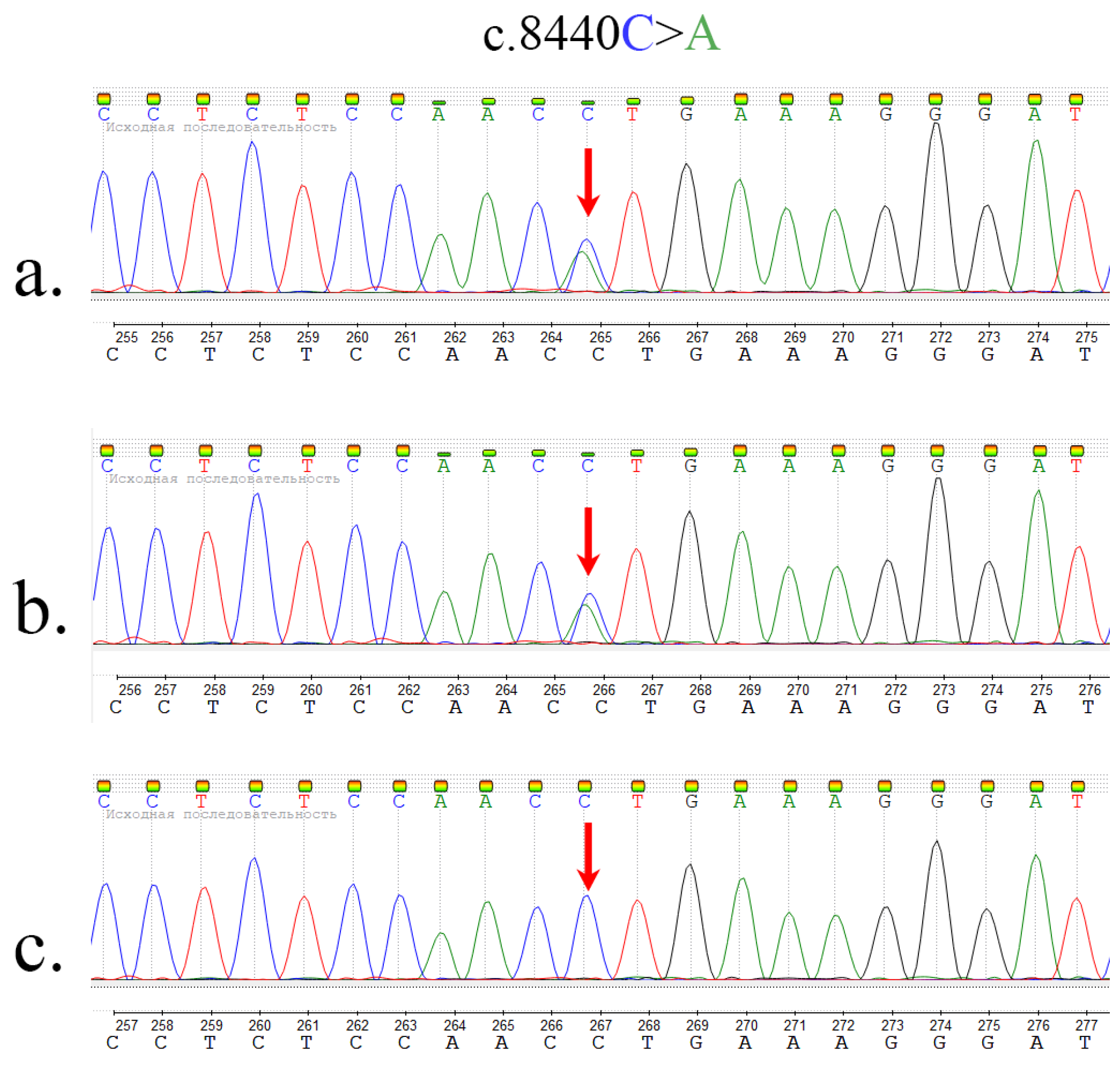
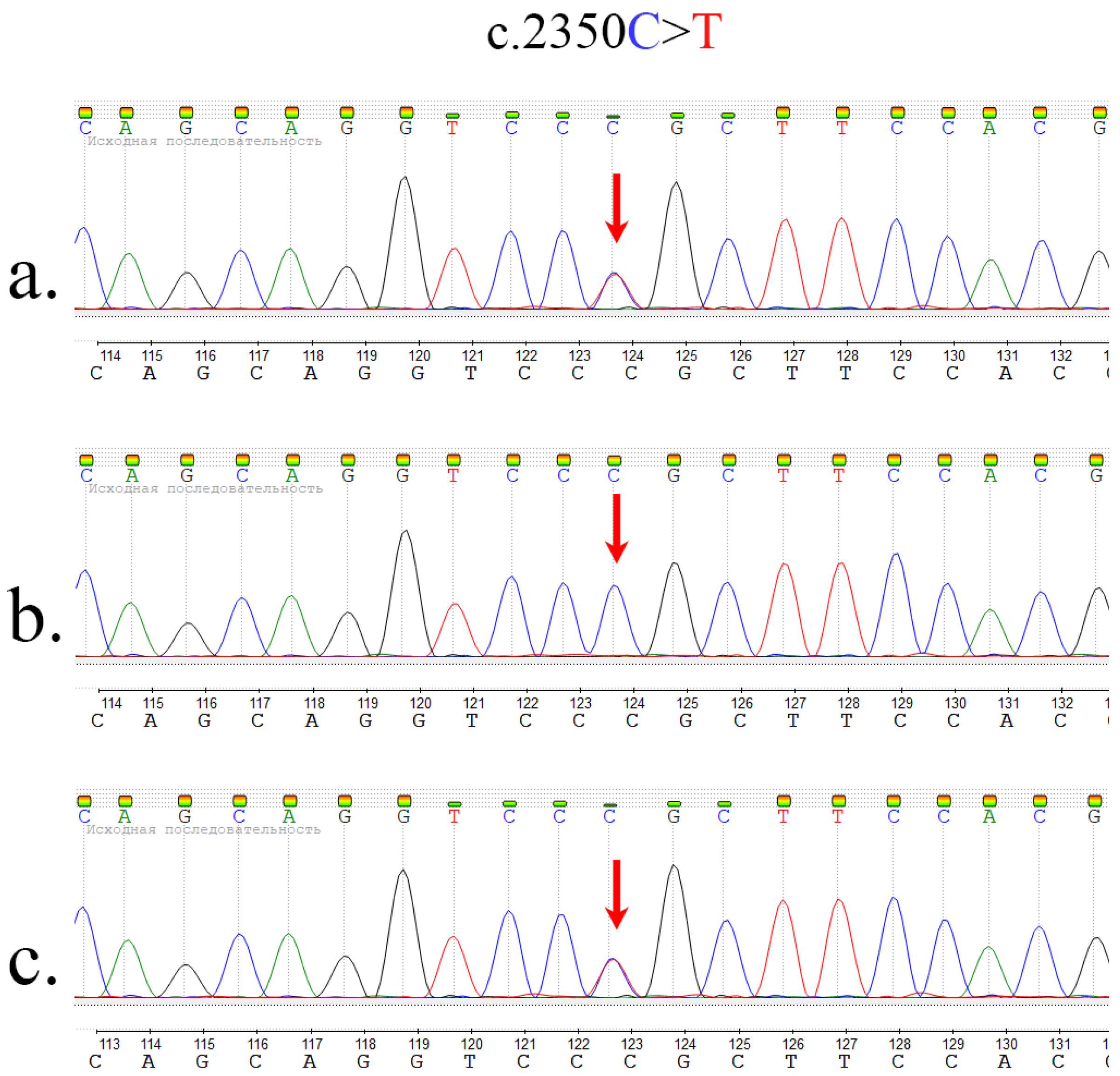
2.2. Whole Exome Sequencing and Sanger Sequencing
3. Discussion
4. Materials and Methods
4.1. Study Participants and Samples Preparations
4.2. Whole Exome Sequencing
4.3. Bioinformatics Data Analysis
4.4. Sanger Sequencing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harjes, P.; Wanker, E.E. The hunt for huntingtin function: Interaction partners tell many different stories. Trends Biochem. Sci. 2003, 28, 425–433. [Google Scholar] [CrossRef]
- Shirasaki, D.; Greiner, E.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.; et al. Network Organization of the Huntingtin Proteomic Interactome in Mammalian Brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Roos, R.A.C. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavitt, B.R.; Kordasiewicz, H.B.; Schobel, S.A. Huntingtin-Lowering Therapies for Huntington Disease: A Review of the Evidence of Potential Benefits and Risks. JAMA Neurol. 2020, 77, 764–772. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- White, J.K.; Auerbach, W.; Duyao, M.P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; MacDonald, M.E. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat. Genet. 1997, 17, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Duyao, M.P.; Auerbach, A.B.; Ryan, A.; Persichetti, F.; Barnes, G.T.; McNeil, S.M.; Ge, P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; et al. Inactivation of the Mouse Huntington’s Disease Gene Homolog Hdh. Science 1995, 269, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.; Barbosa, M.; Ameur, A.; Soares, G.; de Sá, J.; Dias, A.I.; Oliveira, G.; Cabral, P.; Temudo, T.; Calado, E.; et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med Genet. 2016, 53, 190. [Google Scholar] [CrossRef] [Green Version]
- Jung, R.; Lee, Y.; Barker, D.; Correia, K.; Shin, B.; Loupe, J.; Collins, R.L.; Lucente, D.; Ruliera, J.; Gillis, T.; et al. Mutations causing Lopes-Maciel-Rodan syndrome are huntingtin hypomorphs. Hum. Mol. Genet. 2021, 30, 135–148. [Google Scholar] [CrossRef]
- McKeown, C.; Read, A.P.; Dodge, A.; Stecko, O.; Mercer, A.; Harris, R. Wolf-Hirschhorn locus is distal to D4S10 on short arm of chromosome 4. J. Med Genet. 1987, 24, 410. [Google Scholar] [CrossRef]
- Rodan, L.H.; Cohen, J.; Fatemi, A.; Gillis, T.; Lucente, D.; Gusella, J.; Picker, J.D. A novel neurodevelopmental disorder associated with compound heterozygous variants in the huntingtin gene. Eur. J. Hum. Genet. 2016, 24, 1826–1827. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, H.; Li, R.; Sun, S.; Weile, J.; Roth, F.P. Improved pathogenicity prediction for rare human missense variants. Am. J. Hum. Genet. 2021, 108, 1891–1906. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Ryzhkova, O.; Kardymon, O.; Prohorchuk, E.; Konovalov, F.; Maslennikov, A.; Stepanov, V.; Afanasyev, A.; Zaklyazminskaya, E.; Kostareva, A.; Pavlov, A.; et al. Guidelines for the interpretation of massive parallel sequencing variants. Med Genet. 2017, 16, 4–17. [Google Scholar]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- der Auwera, G.A.V.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Cambridge, MA, USA, 2020. [Google Scholar]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Auton, A.; Salcedo, T. The 1000 Genomes Project; Springer: New York, NY, USA, 2015; pp. 71–85. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Barbitoff, Y.A.; Skitchenko, R.K.; Poleshchuk, O.I.; Shikov, A.E.; Serebryakova, E.A.; Nasykhova, Y.A.; Polev, D.E.; Shuvalova, A.R.; Shcherbakova, I.V.; Fedyakov, M.A.; et al. Whole-exome sequencing provides insights into monogenic disease prevalence in Northwest Russia. Mol. Genet. Genom. Med. 2019, 7, e964. [Google Scholar] [CrossRef] [Green Version]
- Shikov, A.E.; Barbitoff, Y.A.; Glotov, A.S.; Danilova, M.M.; Tonyan, Z.N.; Nasykhova, Y.A.; Mikhailova, A.A.; Bespalova, O.N.; Kalinin, R.S.; Mirzorustamova, A.M.; et al. Analysis of the Spectrum of ACE2 Variation Suggests a Possible Influence of Rare and Common Variants on Susceptibility to COVID-19 and Severity of Outcome. Front. Genet. 2020, 11, 551220. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A Database of Human Non-synonymous SNVs and Their Functional Predictions and Annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Position | Exon | cDNA Variant (aa) | Allele Frequency * |
|---|---|---|---|---|
| Lopes F. et al. [9] | chr4:3133374 C>T (GRCh37/hg19) | 16 | c.2108C>T p.Pro703Leu rs768047421 | <0.01% |
| Lopes F. et al. [9] | chr4:3162034 C>T (GRCh37/hg19) | 29 | c.3779C>T p.Thr1260Met rs34315806 | 0.83% |
| Rodan L. et al. [12] | chr4:3177388 G>A (GRCh38) | 34 (intron) | c.4463+1G>A rs1060505027 | N/A |
| Rodan L. et al. [12] | chr4:3229927 T>A (GRCh38) | 60 | c.8156T>A p.Leu2719Gln rs1060505028 | N/A |
| Current study | chr4:3134402 C>T | 17 | c.2350C>T p.Arg784Cys rs375919976 | <0.01% |
| Current study | chr4:3235064 C>A | 61 | c.8440C>A p.Leu2814Met | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koshevaya, Y.S.; Kusakin, A.V.; Buchinskaia, N.V.; Pechnikova, V.V.; Serebryakova, E.A.; Koroteev, A.L.; Glotov, A.S.; Glotov, O.S. Description of the First Registered Case of Lopes–Maciel–Rodan Syndrome in Russia. Int. J. Mol. Sci. 2022, 23, 12437. https://doi.org/10.3390/ijms232012437
Koshevaya YS, Kusakin AV, Buchinskaia NV, Pechnikova VV, Serebryakova EA, Koroteev AL, Glotov AS, Glotov OS. Description of the First Registered Case of Lopes–Maciel–Rodan Syndrome in Russia. International Journal of Molecular Sciences. 2022; 23(20):12437. https://doi.org/10.3390/ijms232012437
Chicago/Turabian StyleKoshevaya, Yuliya S., Aleksey V. Kusakin, Natalia V. Buchinskaia, Valentina V. Pechnikova, Elena A. Serebryakova, Alexander L. Koroteev, Andrey S. Glotov, and Oleg S. Glotov. 2022. "Description of the First Registered Case of Lopes–Maciel–Rodan Syndrome in Russia" International Journal of Molecular Sciences 23, no. 20: 12437. https://doi.org/10.3390/ijms232012437