1. Introduction
Peptide detectability is defined as the probability of detecting a peptide from a standard sample analyzed by a proteomics program [
1]. It is used to measure the relationship between the amount of protein in the sample and the peptide detected. In a shotgun protein assay, proteins in the mixture sample are enzymatically decomposed into peptides, and high-throughput peptide analysis is performed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and other techniques to determine the composition and content of proteins in the sample [
1,
2,
3]. In this process, non-site cleavage of sequences, loss of peptide generated during enzymatic hydrolysis, and other abnormalities may lead to the deviation of the probability of peptide detection [
2], thus restricting the identification and quantitative calculation of proteins. Studies have shown that peptide detectability is crucial to detection, analysis, and differential expression of proteins in proteomics [
4,
5,
6]. Therefore, accurate prediction of peptide detectability helps protein detection and expression analysis provides reference value for the discovery of disease biomarkers and clinical research and thus helps us to more deeply understand cell biology and the underlying mechanisms of human diseases.
There is a strong correlation between amino acids and amino acids in the sequence. Researchers introduce contextual information as the feature representation of the peptide sequence level to better describe the global information of the sequence. Charoenkwan et al. [
7] captured the context information of bitter peptides and used it as a feature, and used BiLSTM and DNN to identify bitter peptides. The results showed that the context information could effectively improve the recognition accuracy of bitter peptides. In peptide detectability, Serrano et al. [
8] regarded amino acids as words, utilized word2vec to calculate the embedding vector of each word, and input the convolutional neural network to predict peptide detectability. Cheng et al. [
9] used transformer and bidirectional gated neural network (BiGRU) to capture the context information of peptides, and improved the prediction performance of peptide detectability. These methods capture the context information of peptides and reflect the global information of peptides better. However, word2vec regards amino acids as words to calculate the embedding vector of each amino acid, but the vector is a fixed value. For the same amino acid, the embedding vector is the same regardless of its context, so it cannot fully reflect the context information of the peptides [
9,
10,
11]. Transformer uses the attention mechanism to capture the association between the target word and other words in the text in order to enhance the target word’s semantic representation, but the target word itself will consist of the semantics of the main parts, leading to the information contained in each word after encoding being more inclined to the meaning of the word itself [
12,
13], which could not obtain high quality context features.
With the continuous development of natural language processing (NLP), the researchers used a new NLP technology-BERT to capture the context information. BERT adopts mask language model (MLM) to strengthen the dependence on context and uses output vector as the semantic representation of the whole sentence, thus integrating the semantic information of each word in the text more “fairly” and solve the problem of self-bias [
14]. It has been successfully applied to protein post-translational modification site prediction [
15], peptide recognition [
7,
16] and gene sequence study [
11,
17,
18]. Therefore, BERT can effectively fuse the information of each amino acid to capture the context information of peptides and enhance the prediction performance of peptide detectability.
There are numerous factors that affect peptide detectability, including sequence information, physicochemical properties [
19,
20,
21,
22], context information derived from sequences, etc. [
8,
9]. Tang et al. [
1] encoded the sequence derived information of peptides and input the feedforward neural network to predict the detection ability of peptides. Li et al. [
19] considered the 292-dimensional physicochemical properties, such as peptide length, constructed artificial neural networks to predict peptide detectability. Guruceaga et al. [
21] screened the 106-dimensional physicochemical properties of peptides, such as molecular weight and theoretical isoelectric point, constructed random forest classifier (RF) for peptide detection. Serrano et al. [
8] extracted context information derived from sequences and input it to a convolutional neural network for feature learning, which improved the prediction accuracy of peptide detectivity. In the above studies, sequence information, physicochemical properties, and context information derived from sequences were used to predict peptide detectivity. However, these methods only considered information at a certain level of peptide, resulting in the simplification of the constructed feature space and the absence of some information. Zhang et al. [
23] considered sequence information, sequence derived pseudo-amino acid composition information, and physicochemical properties, using LightGBM to predict non-classical secreted proteins. Xu et al. [
24] used evolutionary information, the physicochemical properties of proteins, and sequence derived K-spaced amino acid pairs information to predict lysine succinylation sites by support vector machine (SVM), indicating that multivariate representation could make up for the unicity of feature space and effectively improve the accuracy of site prediction. The sequence information, physicochemical properties, and sequence derived information of peptides represent peptides from different perspectives. Considering three kinds of information simultaneously to predict peptide detectability can help the model to learn diversified feature representations, obtain richer feature information of peptide, and produce more reliable prediction results. Therefore, this paper intends to use sequence information, physicochemical properties, and context information derived from sequences to construct a multivariate feature space, enrich the embedded feature representation of peptides, and help the model to better predict peptide detectability.
In classification problems, heterogeneity among information is an important data characteristic of information itself [
25]. Fully considering the differences between different information can help the model fully explore the characteristics of each information representation, and learn the high-quality features of specific information. The sequence information, physicochemical properties and context information describe the peptide from different perspectives. After encoding the information, the data dimension and embedding vector represented by each information are completely different, and there is great heterogeneity among the three types of information. Gao et al. [
26] collected the physicochemical properties and peptide digestibility, and linearly linear fuse these information to obtain 588-dimensional vectors, used RF to predict peptide detectability. Yu et al. [
27] considered the physicochemical properties and sequence information, used same CapsNet network to learn the features of the two types of information, respectively, and thus achieved the prediction of peptide detectability. These methods considered the information of different types of peptides and effectively predicted peptide detectability. However, when linear fusion or the same network is used to process different categories of information, the heterogeneity between different categories of information is not considered, which leads to the captured features losing the unique properties of the corresponding category information [
28,
29] and then affecting the prediction performance of the model. Ensemble deep learning strategy “specialized” a single kind of specific information [
30]. Different deep learning technologies are adopted to learn the representation of specific information, which can capture the high-quality features of specific information, solve the heterogeneity problem among different information, and finally ensemble the classification results of multiple independent deep learning models. It has good fault tolerance and reliability. At present, ensemble deep learning has been successfully applied in biological sequence studies [
31,
32,
33,
34], genome analysis [
35], medication adherence [
36] and other fields, providing strong support for the prediction of peptide detectability.
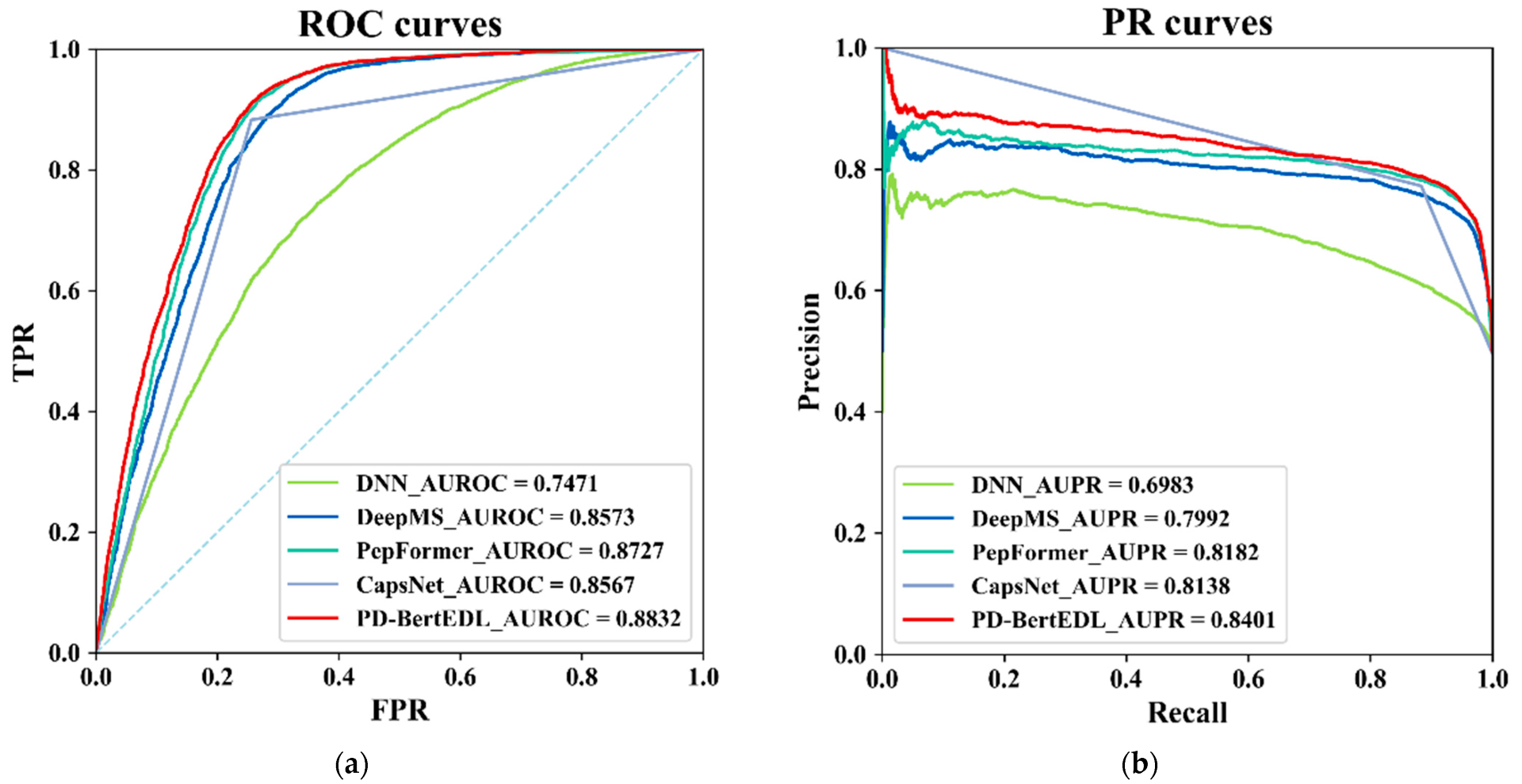
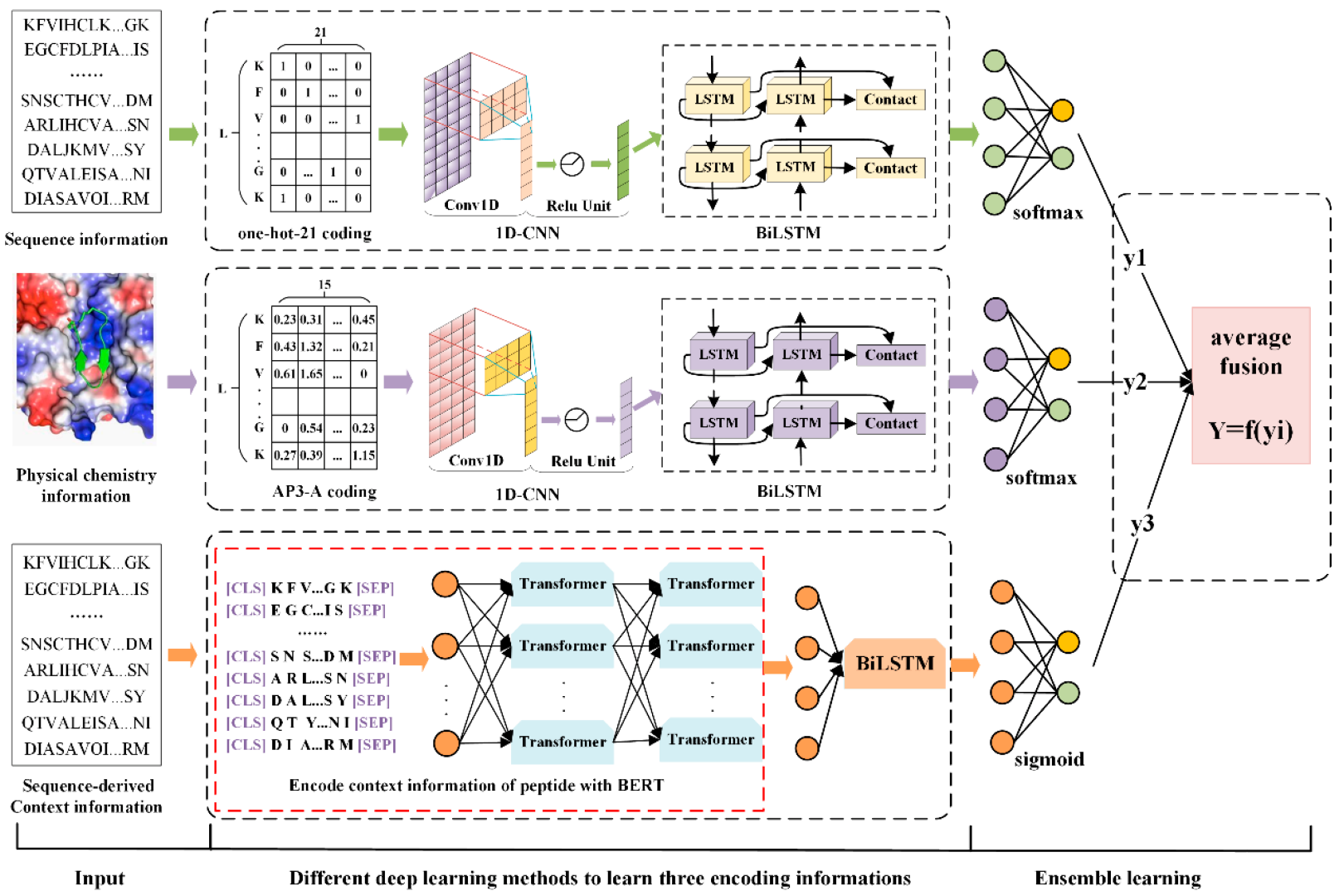
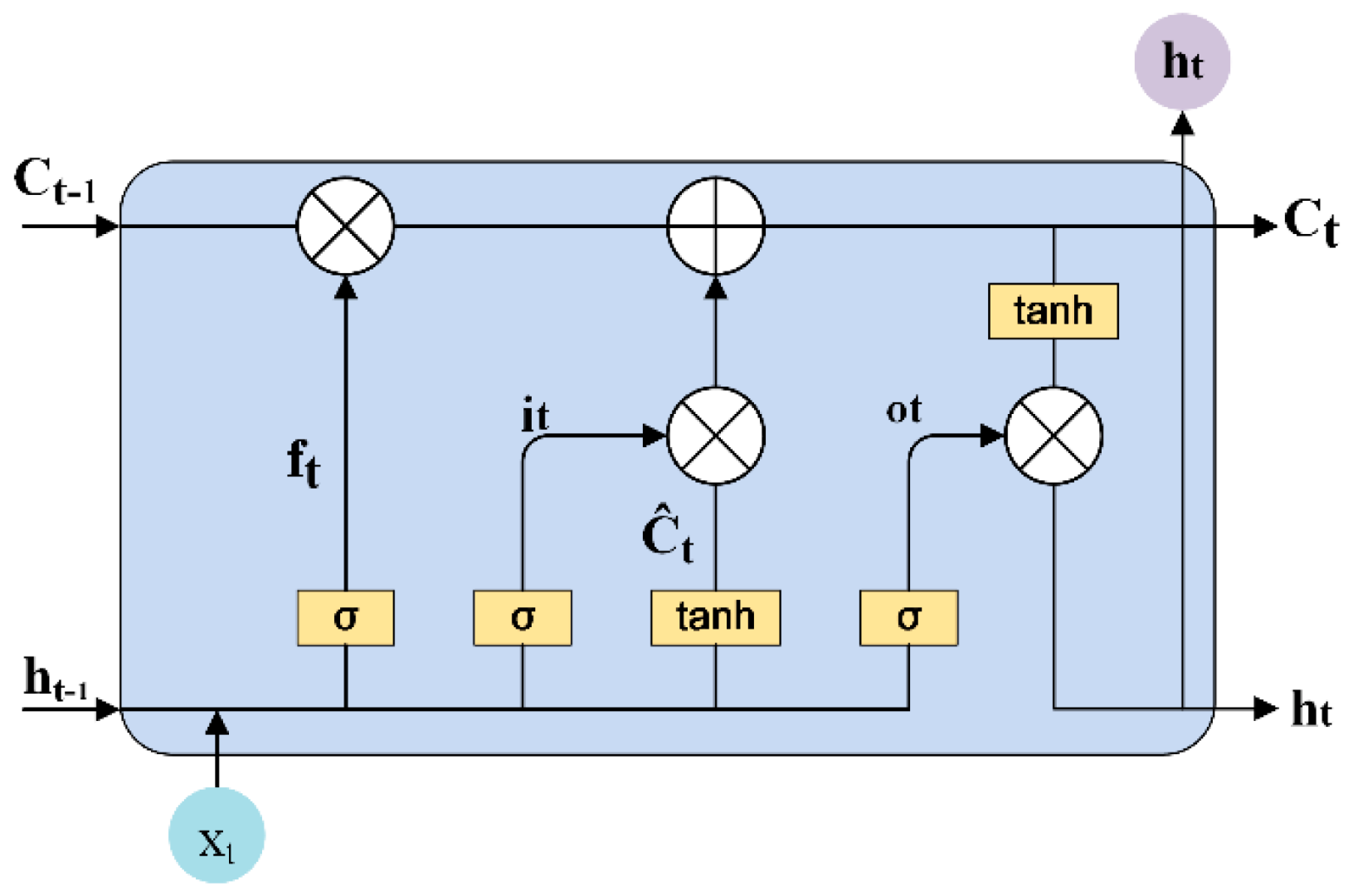
Based on this, we propose PD-BertEDL, an ensemble deep learning method to predict peptide detectability. We introduced the dynamic bidirectional word embedding model BERT to capture the context information of peptides, and combined the context information with the sequence information and physicochemical information of peptides to construct multivariate feature space of peptides. Aiming at three types of different information of peptides, the ensemble deep learning strategy was introduced, and different deep learning methods were selected to capture the high-quality features of specific information: convolutional neural network (CNN) and bidirectional long short-term memory network (BiLSTM) were used to extract the local and global features of sequence information; CNN and BiLSTM were used to study the physicochemical characteristics of amino acids; and BiLSTM was used to learn the context characteristics of the peptides and to describe the global information of the sequence better. Then, the average fusion strategy was utilized to integrate the model prediction results based on the three types of features as the final prediction result to achieve the prediction of peptide detectability.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}