
Insights on Microbial Communities Inhabiting Non-Volcanic Hot Springs
 ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Assessment of the Microbial Community Potential for Retrieving Enzymes of Bio-Technological Interest
2.1.1. Alignment against the SEED Subsystems Database
2.1.2. Alignment to the Non-Redundant Database
2.2. MG-RAST Server Metagenome Overview
2.3. Metabolism and Interactions of the Microbial Community Inferred from the Metagenomic Analysis
2.3.1. Dominant Genera and Main Metabolic Pathways of the Muiño da Veiga Hot Spring Microbial Community
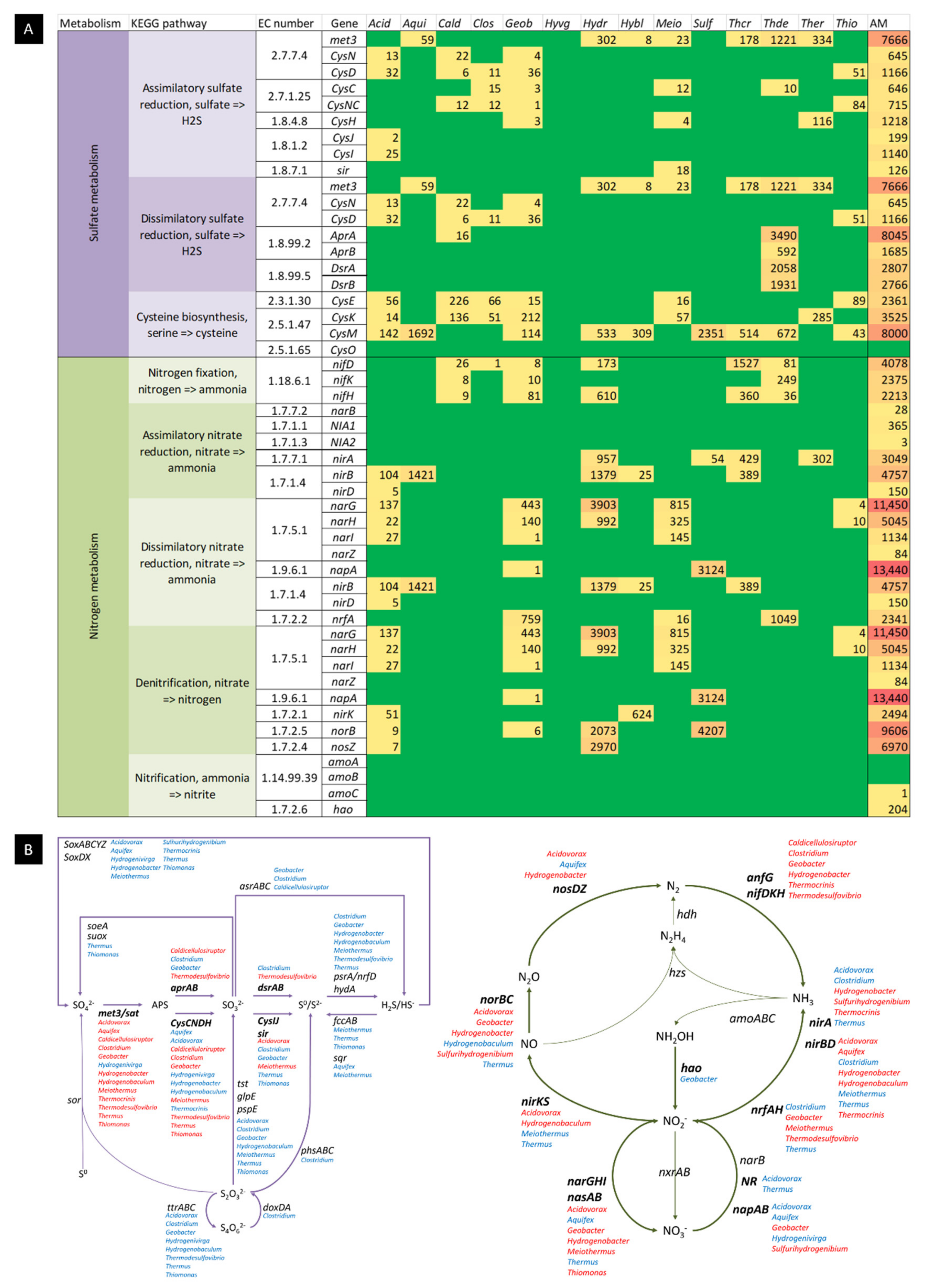
2.3.2. Sulphur Oxidizing Bacteria
2.3.3. Sulphur Reducing Bacteria
2.3.4. Non-Sulphur Metabolism-Related Microorganisms within the Dominant Genera of the Muiño da Veiga Hot Spring
2.3.5. Nitrogen Metabolism
2.3.6. Hydrogen Metabolism
2.3.7. Carbon Metabolism
2.3.8. Rare Species Metabolism
Photosynthesis in Extreme Temperature Conditions
Archaea and Roles in Methane Metabolism
2.4. Assembly of MAGs, Taxonomic Classification, and Functional Annotation
2.4.1. KBase Pipeline Read Processing
2.4.2. MAGs Analysis
3. Materials and Methods
3.1. Metagenomic DNA Source: Site Description and Sample Collection
3.2. DNA Extraction
3.3. DNA Sequencing and Sequence Analysis
3.4. Quality Filtering
3.5. Read Merging
3.6. Alignment to the SEED Subsystems Database
3.7. Alignment to the NCBI nr Database
3.8. Sequence Analysis on the MG-RAST Web-Based Server
3.9. Sequence Analysis on the KBase Web-Based Server
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic Identification and in Situ Detection of Individual Microbial Cells without Cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [CrossRef] [PubMed]
- Staley, J.T.; Konopka, A. Measurement of in Situ Activities of Nonphotosynthetic Microorganisms in Aquatic and Terrestrial Habitats. Annu. Rev. Microbiol. 1985, 39, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, V.; Sørheim, R.; Goksøyr, J. Total Bacterial Diversity in Soil and Sediment Communities—A Review. J. Ind. Microbiol. Biotechnol. 1996, 17, 170–178. [Google Scholar] [CrossRef]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-Generation Sequencing Technologies for Environmental DNA Research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Urbieta, M.S.; Donati, E.R.; Chan, K.-G.; Shahar, S.; Sin, L.L.; Goh, K.M. Thermophiles in the Genomic Era: Biodiversity, Science, and Applications. Biotechnol. Adv. 2015, 33, 633–647. [Google Scholar] [CrossRef]
- Galloway-Peña, J.; Hanson, B. Tools for Analysis of the Microbiome. Dig. Dis. Sci. 2020, 65, 674–685. [Google Scholar] [CrossRef] [Green Version]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular Biological Access to the Chemistry of Unknown Soil Microbes: A New Frontier for Natural Products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef] [Green Version]
- Escuder-Rodríguez, J.-J.; DeCastro, M.-E.; Rodríguez-Belmonte, E.; Becerra, M.; María-Isabel, G.-S. Microbiomes of extreme environments. In Microbiomes of Extreme Environments: Biodiversity and Biotechnological Applications, 1st ed.; Yadav, A.N., Rastegari, A.A., Yadav, N., Eds.; CRC Press: Boca Raton, FL, USA, 2021; pp. 87–105. ISBN 9780429328633. [Google Scholar]
- Colman, D.R.; Lindsay, M.R.; Boyd, E.S. Mixing of Meteoric and Geothermal Fluids Supports Hyperdiverse Chemosynthetic Hydrothermal Communities. Nat. Commun. 2019, 10, 681. [Google Scholar] [CrossRef] [Green Version]
- Cousins, C.R.; Fogel, M.; Bowden, R.; Crawford, I.; Boyce, A.; Cockell, C.; Gunn, M. Biogeochemical Probing of Microbial Communities in a Basalt-Hosted Hot Spring at Kverkfjöll Volcano, Iceland. Geobiology 2018, 16, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Kubo, K.; Knittel, K.; Amann, R.; Fukui, M.; Matsuura, K. Sulfur-Metabolizing Bacterial Populations in Microbial Mats of the Nakabusa Hot Spring, Japan. Syst. Appl. Microbiol. 2011, 34, 293–302. [Google Scholar] [CrossRef]
- Delgado-Outeiriño, I.; Araujo-Nespereira, P.; Cid-Fernández, J.A.; Mejuto, J.C.; Martínez-Carballo, E.; Simal-Gándara, J. Behaviour of Thermal Waters through Granite Rocks Based on Residence Time and Inorganic Pattern. J. Hydrol. 2009, 373, 329–336. [Google Scholar] [CrossRef]
- González-Barreiro, C.; Cancho-Grande, B.; Araujo-Nespereira, P.; Cid-Fernández, J.A.; Simal-Gándara, J. Occurrence of Soluble Organic Compounds in Thermal Waters by Ion Trap Mass Detection. Chemosphere 2009, 75, 34–47. [Google Scholar] [CrossRef]
- López, D.L.; Araujo, P.A.; Outeiriño, I.D.; Cid, J.A.; Astray, G. Geochemical Signatures of the Groundwaters from Ourense Thermal Springs, Galicia, Spain. Sustain. Water Resour. Manag. 2019, 5, 103–116. [Google Scholar] [CrossRef]
- Lin, K.-H.; Liao, B.-Y.; Chang, H.-W.; Huang, S.-W.; Chang, T.-Y.; Yang, C.-Y.; Wang, Y.-B.; Lin, Y.-T.K.; Wu, Y.-W.; Tang, S.-L.; et al. Metabolic Characteristics of Dominant Microbes and Key Rare Species from an Acidic Hot Spring in Taiwan Revealed by Metagenomics. BMC Genom. 2015, 16, 1029. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hatt, J.K.; Tsementzi, D.; Rodriguez-R, L.M.; Ruiz-Pérez, C.A.; Weigand, M.R.; Kizer, H.; Maresca, G.; Krishnan, R.; Poretsky, R.; et al. Quantifying the Importance of the Rare Biosphere for Microbial Community Response to Organic Pollutants in a Freshwater Ecosystem. Appl. Environ. Microbiol. 2017, 83, e03321-16. [Google Scholar] [CrossRef] [Green Version]
- Sahm, K.; John, P.; Nacke, H.; Wemheuer, B.; Grote, R.; Daniel, R.; Antranikian, G. High Abundance of Heterotrophic Prokaryotes in Hydrothermal Springs of the Azores as Revealed by a Network of 16S RRNA Gene-Based Methods. Extremophiles 2013, 17, 649–662. [Google Scholar] [CrossRef]
- Takai, K.; Kobayashi, H.; Nealson, K.H.; Horikoshi, K. Sulfurihydrogenibium Subterraneum Gen. Nov., Sp. Nov., from a Subsurface Hot Aquifer. Int. J. Syst. Evol. Microbiol. 2003, 53, 823–827. [Google Scholar] [CrossRef]
- Nakagawa, S.; Shtaih, Z.; Banta, A.; Beveridge, T.J.; Sako, Y.; Reysenbach, A.-L. Sulfurihydrogenibium Yellowstonense Sp. Nov., an Extremely Thermophilic, Facultatively Heterotrophic, Sulfur-Oxidizing Bacterium from Yellowstone National Park, and Emended Descriptions of the Genus Sulfurihydrogenibium, Sulfurihydrogenibium Subterraneum. Int. J. Syst. Evol. Microbiol. 2005, 55, 2263–2268. [Google Scholar] [CrossRef]
- Takacs-Vesbach, C.; Inskeep, W.P.; Jay, Z.J.; Herrgard, M.J.; Rusch, D.B.; Tringe, S.G.; Kozubal, M.A.; Hamamura, N.; Macur, R.E.; Fouke, B.W.; et al. Metagenome Sequence Analysis of Filamentous Microbial Communities Obtained from Geochemically Distinct Geothermal Channels Reveals Specialization of Three Aquificales Lineages. Front. Microbiol. 2013, 4, 84. [Google Scholar] [CrossRef] [Green Version]
- Dodsworth, J.A.; Hungate, B.A.; Hedlund, B.P. Ammonia Oxidation, Denitrification and Dissimilatory Nitrate Reduction to Ammonium in Two US Great Basin Hot Springs with Abundant Ammonia-Oxidizing Archaea. Environ. Microbiol. 2011, 13, 2371–2386. [Google Scholar] [CrossRef]
- Huber, R.; Wilharm, T.; Huber, D.; Trincone, A.; Burggraf, S.; König, H.; Reinhard, R.; Rockinger, I.; Fricke, H.; Stetter, K.O. Aquifex Pyrophilus Gen. Nov. Sp. Nov., Represents a Novel Group of Marine Hyperthermophilic Hydrogen-Oxidizing Bacteria. Syst. Appl. Microbiol. 1992, 15, 340–351. [Google Scholar] [CrossRef]
- Deckert, G.; Warren, P.V.; Gaasterland, T.; Young, W.G.; Lenox, A.L.; Graham, D.E.; Overbeek, R.; Snead, M.A.; Keller, M.; Aujay, M.; et al. The Complete Genome of the Hyperthermophilic Bacterium Aquifex Aeolicus. Nature 1998, 392, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Beh, M.; Strauss, G.; Huber, R.; Stetter, K.-O.; Fuchs, G. Enzymes of the Reductive Citric Acid Cycle in the Autotrophic Eubacterium Aquifex Pyrophilus and in the Archaebacterium Thermoproteus Neutrophilus. Arch. Microbiol. 1993, 160, 306–311. [Google Scholar] [CrossRef]
- Bonjour, F.; Aragno, M. Growth of Thermophilic, Obligatorily Chemolithoautotrophic Hydrogen-Oxidizing Bacteria Related to Hydrogenobacter with Thiosulfate and Elemental Sulfur as Electron and Energy Source. FEMS Microbiol. Lett. 1986, 35, 11–15. [Google Scholar] [CrossRef]
- Alfredsson, G.A.; Ingason, A.; Kristjansson, J.K. Growth of Thermophilic Obligately Autotrophic Hydrogen-Oxidizing Bacteria on Thiosulfate. Lett. Appl. Microbiol. 1986, 2, 21–23. [Google Scholar] [CrossRef]
- Shiba, H.; Kawasumi, T.; Igarashi, Y.; Kodama, T.; Minoda, Y. The CO2 Assimilation via the Reductive Tricarboxylic Acid Cycle in an Obligately Autotrophic, Aerobic Hydrogen-Oxidizing Bacterium, Hydrogenobacter Thermophilus. Arch. Microbiol. 1985, 141, 198–203. [Google Scholar] [CrossRef]
- Kawasumi, T.; Igarashi, Y.; Kodama, T.; Minoda, Y. Hydrogenobacter Thermophilus Gen. Nov., Sp. Nov., an Extremely Thermophilic, Aerobic, Hydrogen-Oxidizing Bacterium. Int. J. Syst. Bacteriol. 1984, 34, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Pitulle, C.; Yang, Y.; Marchiani, M.; Moore, E.R.B.; Siefert, J.L.; Aragno, M.; Jurtshuk, P.; Fox, G.E. Phylogenetic Position of the Genus Hydrogenobacter. Int. J. Syst. Bacteriol. 1994, 44, 620–626. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.; Eder, W.; Heldwein, S.; Wanner, G.; Huber, H.; Rachel, R.; Stetter, K.O. Thermocrinis Ruber Gen. Nov., Sp. Nov., a Pink-Filament-Forming Hyperthermophilic Bacterium Isolated from Yellowstone National Park. Appl. Environ. Microbiol. 1998, 64, 3576–3583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodsworth, J.A.; Ong, J.C.; Williams, A.J.; Dohnalkova, A.C.; Hedlund, B.P. Thermocrinis Jamiesonii Sp. Nov., a Thiosulfate-Oxidizing, Autotropic Thermophile Isolated from a Geothermal Spring. Int. J. Syst. Evol. Microbiol. 2015, 65, 4769–4775. [Google Scholar] [CrossRef] [PubMed]
- Eder, W.; Huber, R. New Isolates and Physiological Properties of the Aquificales and Description of Thermocrinis Albus Sp. Nov. Extremophiles 2002, 6, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Härtig, C.; Lohmayer, R.; Kolb, S.; Horn, M.A.; Inskeep, W.P.; Planer-Friedrich, B. Chemolithotrophic Growth of the Aerobic Hyperthermophilic Bacterium Thermocrinis Ruber OC 14/7/2 on Monothioarsenate and Arsenite. FEMS Microbiol. Ecol. 2014, 90, 747–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, S.L.; Liu, Y.; Ferrera, I.; Beveridge, T.; Reysenbach, A.-L. Thermocrinis Minervae Sp. Nov., a Hydrogen- and Sulfur-Oxidizing, Thermophilic Member of the Aquificales from a Costa Rican Terrestrial Hot Spring. Int. J. Syst. Evol. Microbiol. 2010, 60, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Massello, F.L.; Chan, C.S.; Chan, K.-G.; Goh, K.M.; Donati, E.; Urbieta, M.S. Meta-Analysis of Microbial Communities in Hot Springs: Recurrent Taxa and Complex Shaping Factors beyond PH and Temperature. Microorganisms 2020, 8, 906. [Google Scholar] [CrossRef]
- Stohr, R.; Waberski, A.; Völker, H.; Tindall, B.J.; Thomm, M. Hydrogenothermus Marinus Gen. Nov., Sp. Nov., a Novel Thermophilic Hydrogen-Oxidizing Bacterium, Recognition of Calderobacterium Hydrogenophilum as a Member of the Genus Hydrogenobacter and Proposal of the Reclassification of Hydrogenobacter Acidophilus a. Int. J. Syst. Evol. Microbiol. 2001, 51, 1853–1862. [Google Scholar] [CrossRef]
- Romano, C.; D’Imperio, S.; Woyke, T.; Mavromatis, K.; Lasken, R.; Shock, E.L.; McDermott, T.R. Comparative Genomic Analysis of Phylogenetically Closely Related Hydrogenobaculum Sp. Isolates from Yellowstone National Park. Appl. Environ. Microbiol. 2013, 79, 2932–2943. [Google Scholar] [CrossRef] [Green Version]
- Hedlund, B.P.; Reysenbach, A.-L.; Huang, L.; Ong, J.C.; Liu, Z.; Dodsworth, J.A.; Ahmed, R.; Williams, A.J.; Briggs, B.R.; Liu, Y.; et al. Isolation of Diverse Members of the Aquificales from Geothermal Springs in Tengchong, China. Front. Microbiol. 2015, 6, 157. [Google Scholar] [CrossRef]
- Donahoe-Christiansen, J.; D’Imperio, S.; Jackson, C.R.; Inskeep, W.P.; McDermott, T.R. Arsenite-Oxidizing Hydrogenobaculum Strain Isolated from an Acid-Sulfate-Chloride Geothermal Spring in Yellowstone National Park. Appl. Environ. Microbiol. 2004, 70, 1865–1868. [Google Scholar] [CrossRef] [Green Version]
- Moreira, D.; Amils, R. Phylogeny of Thiobacillus Cuprinus and Other Mixotrophic Thiobacilli: Proposal for Thiomonas Gen. Nov. Int. J. Syst. Bacteriol. 1997, 47, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Vésteinsdóttir, H.; Reynisdóttir, D.B.; Örlygsson, J. Thiomonas Islandica Sp. Nov., a Moderately Thermophilic, Hydrogen- and Sulfur-Oxidizing Betaproteobacterium Isolated from a Hot Spring. Int. J. Syst. Evol. Microbiol. 2011, 61, 132–137. [Google Scholar] [CrossRef]
- Bryan, C.G.; Marchal, M.; Battaglia-Brunet, F.; Kugler, V.; Lemaitre-Guillier, C.; Lièvremont, D.; Bertin, P.N.; Arsène-Ploetze, F. Carbon and Arsenic Metabolism in Thiomonas Strains: Differences Revealed Diverse Adaptation Processes. BMC Microbiol. 2009, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.; Nakamura, S.; Inagaki, F.; Takai, K.; Shirai, N.; Sako, Y. Hydrogenivirga Caldilitoris Gen. Nov., Sp. Nov., a Novel Extremely Thermophilic, Hydrogen- and Sulfur-Oxidizing Bacterium from a Coastal Hydrothermal Field. Int. J. Syst. Evol. Microbiol. 2004, 54, 2079–2084. [Google Scholar] [CrossRef] [Green Version]
- Nunoura, T.; Miyazaki, M.; Suzuki, Y.; Takai, K.; Horikoshi, K. Hydrogenivirga Okinawensis Sp. Nov., a Thermophilic Sulfur-Oxidizing Chemolithoautotroph Isolated from a Deep-Sea Hydrothermal Field, Southern Okinawa Trough. Int. J. Syst. Evol. Microbiol. 2008, 58, 676–681. [Google Scholar] [CrossRef]
- Henry, E.A.; Devereux, R.; Maki, J.S.; Gilmour, C.C.; Woese, C.R.; Mandelco, L.; Schauder, R.; Remsen, C.C.; Mitchell, R. Characterization of a New Thermophilic Sulfate-Reducing Bacterium. Arch. Microbiol. 1994, 161, 62–69. [Google Scholar] [CrossRef]
- Lovley, D.R.; Giovannoni, S.J.; White, D.C.; Champine, J.E.; Phillips, E.J.P.; Gorby, Y.A.; Goodwin, S. Geobacter Metallireducens Gen. Nov. Sp. Nov., a Microorganism Capable of Coupling the Complete Oxidation of Organic Compounds to the Reduction of Iron and Other Metals. Arch. Microbiol. 1993, 159, 336–344. [Google Scholar] [CrossRef]
- Caccavo, F.; Lonergan, D.J.; Lovley, D.R.; Davis, M.; Stolz, J.F.; McInerney, M.J. Geobacter Sulfurreducens Sp. Nov., a Hydrogen- and Acetate-Oxidizing Dissimilatory Metal-Reducing Microorganism. Appl. Environ. Microbiol. 1994, 60, 3752–3759. [Google Scholar] [CrossRef] [Green Version]
- Fortney, N.W.; He, S.; Converse, B.J.; Beard, B.L.; Johnson, C.M.; Boyd, E.S.; Roden, E.E. Microbial Fe(III) Oxide Reduction Potential in Chocolate Pots Hot Spring, Yellowstone National Park. Geobiology 2016, 14, 255–275. [Google Scholar] [CrossRef] [Green Version]
- Sekiguchi, Y.; Muramatsu, M.; Imachi, H.; Narihiro, T.; Ohashi, A.; Harada, H.; Hanada, S.; Kamagata, Y. Thermodesulfovibrio Aggregans Sp. Nov. and Thermodesulfovibrio Thiophilus Sp. Nov., Anaerobic, Thermophilic, Sulfate-Reducing Bacteria Isolated from Thermophilic Methanogenic Sludge, and Emended Description of the Genus Thermodesulfovibrio. Int. J. Syst. Evol. Microbiol. 2008, 58, 2541–2548. [Google Scholar] [CrossRef] [Green Version]
- Zhou, E.-M.; Adegboruwa, A.L.; Mefferd, C.C.; Bhute, S.S.; Murugapiran, S.K.; Dodsworth, J.A.; Thomas, S.C.; Bengtson, A.J.; Liu, L.; Xian, W.-D.; et al. Diverse Respiratory Capacity among Thermus Strains from US Great Basin Hot Springs. Extremophiles 2020, 24, 71–80. [Google Scholar] [CrossRef]
- Balkwill, D.L.; Kieft, T.L.; Tsukuda, T.; Kostandarithes, H.M.; Onstott, T.C.; Bownas, J.; Macnaughton, S.; Fredrickson, J.K. Identification of Iron-Reducing Thermus Strains as Thermus Scotoductus. Extremophiles 2004, 8, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.; Falsen, E.; Pot, B.; Jantzen, E.; Hoste, B.; Vandamme, P.; Gillis, M.; Kersters, K.; De Ley, J. Acidovorax, a New Genus for Pseudomonas Facilis, Pseudomonas Delafieldii, E. Falsen (EF) Group 13, EF Group 16, and Several Clinical Isolates, with the Species Acidovorax Facilis Comb. Nov., Acidovorax Delafieldii Comb. Nov., and Acidovorax Temperans sp. Int. J. Syst. Bacteriol. 1990, 40, 384–398. [Google Scholar] [CrossRef]
- Saxena, R.; Dhakan, D.B.; Mittal, P.; Waiker, P.; Chowdhury, A.; Ghatak, A.; Sharma, V.K. Metagenomic Analysis of Hot Springs in Central India Reveals Hydrocarbon Degrading Thermophiles and Pathways Essential for Survival in Extreme Environments. Front. Microbiol. 2017, 7, 2123. [Google Scholar] [CrossRef]
- Padilla-Del Valle, R.; Morales-Vale, L.R.; Ríos-Velázquez, C. Unraveling the Microbial and Functional Diversity of Coamo Thermal Spring in Puerto Rico Using Metagenomic Library Generation and Shotgun Sequencing. Genom. Data 2017, 11, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Kozubal, M.A.; Macur, R.E.; Jay, Z.J.; Beam, J.P.; Malfatti, S.A.; Tringe, S.G.; Kocar, B.D.; Borch, T.; Inskeep, W.P. Microbial Iron Cycling in Acidic Geothermal Springs of Yellowstone National Park: Integrating Molecular Surveys, Geochemical Processes, and Isolation of Novel Fe-Active Microorganisms. Front. Microbiol. 2012, 3, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvarajan, R.; Sibanda, T.; Tekere, M. Thermophilic Bacterial Communities Inhabiting the Microbial Mats of “Indifferent” and Chalybeate (Iron-Rich) Thermal Springs: Diversity and Biotechnological Analysis. Microbiologyopen 2018, 7, e00560. [Google Scholar] [CrossRef] [PubMed]
- Heylen, K.; Lebbe, L.; De Vos, P. Acidovorax Caeni Sp. Nov., a Denitrifying Species with Genetically Diverse Isolates from Activated Sludge. Int. J. Syst. Evol. Microbiol. 2008, 58, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Udaondo, Z.; Duque, E.; Ramos, J.-L. The Pangenome of the Genus Clostridium. Environ. Microbiol. 2017, 19, 2588–2603. [Google Scholar] [CrossRef]
- Liu, L.; Jiao, J.-Y.; Fang, B.-Z.; Lv, A.-P.; Ming, Y.-Z.; Li, M.-M.; Salam, N.; Li, W.-J. Isolation of Clostridium from Yunnan-Tibet Hot Springs and Description of Clostridium Thermarum Sp. Nov. with Lignocellulosic Ethanol Production. Syst. Appl. Microbiol. 2020, 43, 126104. [Google Scholar] [CrossRef]
- Rainey, F.A.; Donnison, A.M.; Janssen, P.H.; Saul, D.; Rodrigo, A.; Bergquist, P.L.; Daniel, R.M.; Stackebrandt, E.; Morgan, H.W. Description of Caldicellulosiruptor Saccharolyticus Gen. Nov., Sp. Nov: An Obligately Anaerobic, Extremely Thermophilic, Cellulolytic Bacterium. FEMS Microbiol. Lett. 1994, 120, 263–266. [Google Scholar] [CrossRef]
- Blumer-Schuette, S.E.; Giannone, R.J.; Zurawski, J.V.; Ozdemir, I.; Ma, Q.; Yin, Y.; Xu, Y.; Kataeva, I.; Poole, F.L.; Adams, M.W.W.; et al. Caldicellulosiruptor Core and Pangenomes Reveal Determinants for Noncellulosomal Thermophilic Deconstruction of Plant Biomass. J. Bacteriol. 2012, 194, 4015–4028. [Google Scholar] [CrossRef]
- Bellini, M.I.; Gutiérrez, L.; Tarlera, S.; Scavino, A.F. Isolation and Functional Analysis of Denitrifiers in an Aquifer with High Potential for Denitrification. Syst. Appl. Microbiol. 2013, 36, 505–516. [Google Scholar] [CrossRef]
- Zeytun, A.; Sikorski, J.; Nolan, M.; Lapidus, A.; Lucas, S.; Han, J.; Tice, H.; Cheng, J.-F.; Tapia, R.; Goodwin, L.; et al. Complete Genome Sequence of Hydrogenobacter Thermophilus Type Strain (TK-6T). Stand. Genom. Sci. 2011, 4, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, A.; Thiel, V.; Matsuura, K.; McGlynn, S.E.; Haruta, S. Phylogenetic Diversity of Nitrogenase Reductase Genes and Possible Nitrogen-Fixing Bacteria in Thermophilic Chemosynthetic Microbial Communities in Nakabusa Hot Springs. Microbes Environ. 2018, 33, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, T.L.; Koonce, E.; Howells, A.; Havig, J.R.; Jewell, T.; de la Torre, J.R.; Peters, J.W.; Boyd, E.S. Competition for Ammonia Influences the Structure of Chemotrophic Communities in Geothermal Springs. Appl. Environ. Microbiol. 2014, 80, 653–661. [Google Scholar] [CrossRef] [Green Version]
- Pester, M.; Rattei, T.; Flechl, S.; Gröngröft, A.; Richter, A.; Overmann, J.; Reinhold-Hurek, B.; Loy, A.; Wagner, M. AmoA-based Consensus Phylogeny of Ammonia-oxidizing Archaea and Deep Sequencing of AmoA Genes from Soils of Four Different Geographic Regions. Environ. Microbiol. 2012, 14, 525–539. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.L.; Ye, Q.; Huang, Z.; Li, W.; Chen, J.; Song, Z.; Zhao, W.; Bagwell, C.; Inskeep, W.P.; Ross, C.; et al. Global Occurrence of Archaeal AmoA Genes in Terrestrial Hot Springs. Appl. Environ. Microbiol. 2008, 74, 6417–6426. [Google Scholar] [CrossRef] [Green Version]
- Reigstad, L.J.; Richter, A.; Daims, H.; Urich, T.; Schwark, L.; Schleper, C. Nitrification in Terrestrial Hot Springs of Iceland and Kamchatka. FEMS Microbiol. Ecol. 2008, 64, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Revsbech, N.P.; Trampe, E.; Lichtenberg, M.; Ward, D.M.; Kühl, M. In Situ Hydrogen Dynamics in a Hot Spring Microbial Mat during a Diel Cycle. Appl. Environ. Microbiol. 2016, 82, 4209–4217. [Google Scholar] [CrossRef] [Green Version]
- Otaki, H.; Everroad, R.C.; Matsuura, K.; Haruta, S. Production and Consumption of Hydrogen in Hot Spring Microbial Mats Dominated by a Filamentous Anoxygenic Photosynthetic Bacterium. Microbes Environ. 2012, 27, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, M.R.; Colman, D.R.; Amenabar, M.J.; Fristad, K.E.; Fecteau, K.M.; Debes, R.V.; Spear, J.R.; Shock, E.L.; Hoehler, T.M.; Boyd, E.S. Probing the Geological Source and Biological Fate of Hydrogen in Yellowstone Hot Springs. Environ. Microbiol. 2019, 21, 3816–3830. [Google Scholar] [CrossRef]
- Fenchel, T.; King, G.M.; Blackburn, T.H. Bacterial Metabolism. In Bacterial Biogeochemistry; Elsevier: Amsterdam, The Netherlands, 2012; pp. 1–34. ISBN 9780124158368. [Google Scholar]
- Lynch, M.D.J.; Neufeld, J.D. Ecology and Exploration of the Rare Biosphere. Nat. Rev. Microbiol. 2015, 13, 217–229. [Google Scholar] [CrossRef]
- Yang, S.; Winkel, M.; Wagner, D.; Liebner, S. Community Structure of Rare Methanogenic Archaea: Insight from a Single Functional Group. FEMS Microbiol. Ecol. 2017, 93, fix126. [Google Scholar] [CrossRef] [Green Version]
- Boyd, E.S.; Leavitt, W.D.; Geesey, G.G. CO2 Uptake and Fixation by a Thermoacidophilic Microbial Community Attached to Precipitated Sulfur in a Geothermal Spring. Appl. Environ. Microbiol. 2009, 75, 4289–4296. [Google Scholar] [CrossRef] [Green Version]
- Bennett, A.C.; Murugapiran, S.K.; Hamilton, T.L. Temperature Impacts Community Structure and Function of Phototrophic Chloroflexi and Cyanobacteria in Two Alkaline Hot Springs in Yellowstone National Park. Environ. Microbiol. Rep. 2020, 12, 503–513. [Google Scholar] [CrossRef]
- Maeder, D.L.; Anderson, I.; Brettin, T.S.; Bruce, D.C.; Gilna, P.; Han, C.S.; Lapidus, A.; Metcalf, W.W.; Saunders, E.; Tapia, R.; et al. The Methanosarcina Barkeri Genome: Comparative Analysis with Methanosarcina Acetivorans and Methanosarcina Mazei Reveals Extensive Rearrangement within Methanosarcinal Genomes. J. Bacteriol. 2006, 188, 7922–7931. [Google Scholar] [CrossRef] [Green Version]
- Huber, H.; Thomm, M.; König, H.; Thies, G.; Stetter, K.O. Methanococcus Thermolithotrophicus, a Novel Thermophilic Lithotrophic Methanogen. Arch. Microbiol. 1982, 132, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Jones, W.J.; Leigh, J.A.; Mayer, F.; Woese, C.R.; Wolfe, R.S. Methanococcus Jannaschii Sp. Nov., an Extremely Thermophilic Methanogen from a Submarine Hydrothermal Vent. Arch. Microbiol. 1983, 136, 254–261. [Google Scholar] [CrossRef]
- Smith, K.S.; Ingram-Smith, C. Methanosaeta, the Forgotten Methanogen? Trends Microbiol. 2007, 15, 150–155. [Google Scholar] [CrossRef]
- Liu, C.; Sun, D.; Zhao, Z.; Dang, Y.; Holmes, D.E. Methanothrix Enhances Biogas Upgrading in Microbial Electrolysis Cell via Direct Electron Transfer. Bioresour. Technol. 2019, 291, 121877. [Google Scholar] [CrossRef]
- Wasserfallen, A.; Nölling, J.; Pfister, P.; Reeve, J.; Conway de Macario, E. Phylogenetic Analysis of 18 Thermophilic Methanobacterium Isolates Supports the Proposals to Create a New Genus, Methanothermobacter Gen. Nov., and to Reclassify Several Isolates in Three Species, Methanothermobacter Thermautotrophicus Comb. nov., Methanothermobacter wolfeii comb. nov., and Methanothermobacter marburgensis sp. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Stetter, K.O. Archaeoglobus Fulgidus Gen. Nov., Sp. Nov.: A New Taxon of Extremely Thermophilic Archaebacteria. Syst. Appl. Microbiol. 1988, 10, 172–173. [Google Scholar] [CrossRef]
- Callac, N.; Oger, P.; Lesongeur, F.; Rattray, J.E.; Vannier, P.; Michoud, G.; Beauverger, M.; Gayet, N.; Rouxel, O.; Jebbar, M.; et al. Pyrococcus Kukulkanii Sp. Nov., a Hyperthermophilic, Piezophilic Archaeon Isolated from a Deep-Sea Hydrothermal Vent. Int. J. Syst. Evol. Microbiol. 2016, 66, 3142–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmasso, C.; Oger, P.; Selva, G.; Courtine, D.; L’Haridon, S.; Garlaschelli, A.; Roussel, E.; Miyazaki, J.; Reveillaud, J.; Jebbar, M.; et al. Thermococcus Piezophilus Sp. Nov., a Novel Hyperthermophilic and Piezophilic Archaeon with a Broad Pressure Range for Growth, Isolated from a Deepest Hydrothermal Vent at the Mid-Cayman Rise. Syst. Appl. Microbiol. 2016, 39, 440–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, G.; Spinnler, C.; Gambacorta, A.; Stetter, K.O. Metallosphaera Sedula Gen. and Sp. Nov. Represents a New Genus of Aerobic, Metal-Mobilizing, Thermoacidophilic Archaebacteria. Syst. Appl. Microbiol. 1989, 12, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Miroshnichenko, M.L.; Lebedinsky, A.V.; Chernyh, N.A.; Tourova, T.P.; Kolganova, T.V.; Spring, S.; Bonch-Osmolovskaya, E.A. Caldimicrobium Rimae Gen. Nov., Sp. Nov., an Extremely Thermophilic, Facultatively Lithoautotrophic, Anaerobic Bacterium from the Uzon Caldera, Kamchatka. Int. J. Syst. Evol. Microbiol. 2009, 59, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, H.; Umezawa, K.; Fukui, M. Caldimicrobium ThioDis.mutans Sp. Nov., a Sulfur-Disproportionating Bacterium Isolated from a Hot Spring, and Emended Description of the Genus Caldimicrobium. Int. J. Syst. Evol. Microbiol. 2016, 66, 1828–1831. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Tang, R.; Yu, S.; Zheng, B.; Feng, Y. A Novel Thermostable Cellulase from Fervidobacterium Nodosum. J. Mol. Catal. B Enzym. 2010, 66, 294–301. [Google Scholar] [CrossRef]
- Morris, D.D.; Gibbs, M.D.; Chin, C.W.J.; Koh, M.-H.; Wong, K.K.Y.; Allison, R.W.; Nelson, P.J.; Bergquist, P.L. Cloning of the XynB Gene from Dictyoglomus Thermophilum Rt46B.1 and Action of the Gene Product on Kraft Pulp. Appl. Environ. Microbiol. 1998, 64, 1759–1765. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Li, Z.; Ye, Q.; Xu, J.; Liu, Y. Heterologous Expression and Characterization of a Novel Thermo-Halotolerant Endoglucanase Cel5H from Dictyoglomus Thermophilum. Bioresour. Technol. 2013, 142, 338–344. [Google Scholar] [CrossRef]
- Bretagne, D.; Pâris, A.; de Vaumas, R.; Lafite, P.; Daniellou, R. Crystal Structure of Dictyoglomus Thermophilum β-d-Xylosidase DtXyl Unravels the Structural Determinants for Efficient Notoginsenoside R1 Hydrolysis. Biochimie 2021, 181, 34–41. [Google Scholar] [CrossRef]
- Iino, T.; Nakagawa, T.; Mori, K.; Harayama, S.; Suzuki, K.-i. Calditerrivibrio Nitroreducens Gen. Nov., Sp. Nov., a Thermophilic, Nitrate-Reducing Bacterium Isolated from a Terrestrial Hot Spring in Japan. Int. J. Syst. Evol. Microbiol. 2008, 58, 1675–1679. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of Nearly 8,000 Metagenome-Assembled Genomes Substantially Expands the Tree of Life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The Metagenomics RAST Server—A Public Resource for the Automatic Phylogenetic and Functional Analysis of Metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A Fast and Accurate Illumina Paired-End ReAd MergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A Tool for Agile Functional Analysis of Shotgun Metagenomic Data. Bioinformatics 2016, 32, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- National Library of Medicine US National Center for Biotechnology Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 28 September 2016).
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.-Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The Subsystems Approach to Genome Annotation and Its Use in the Project to Annotate 1000 Genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 September 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.-A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A Complete Domain-to-Species Taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A Standardized Bacterial Taxonomy Based on Genome Phylogeny Substantially Revises the Tree of Life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. Pplacer: Linear Time Maximum-Likelihood and Bayesian Phylogenetic Placement of Sequences onto a Fixed Reference Tree. BMC Bioinform. 2010, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of Microbial Genomes Using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A Modular and Extensible Implementation of the RAST Algorithm for Building Custom Annotation Pipelines and Annotating Batches of Genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Anda, D.; Makk, J.; Krett, G.; Jurecska, L.; Márialigeti, K.; Mádl-Szőnyi, J.; Borsodi, A.K. Thermophilic Prokaryotic Communities Inhabiting the Biofilm and Well Water of a Thermal Karst System Located in Budapest (Hungary). Extremophiles 2015, 19, 787–797. [Google Scholar] [CrossRef]
- Castelán-Sánchez, H.G.; Meza-Rodríguez, P.M.; Carrillo, E.; Ríos-Vázquez, D.I.; Liñan-Torres, A.; Batista-García, R.A.; Pérez-Rueda, E.; Rojas-Ruíz, N.E.; Dávila-Ramos, S. The Microbial Composition in Circumneutral Thermal Springs from Chignahuapan, Puebla, Mexico Reveals the Presence of Particular Sulfur-Oxidizing Bacterial and Viral Communities. Microorganisms 2020, 8, 1677. [Google Scholar] [CrossRef]
- Everroad, R.C.; Otaki, H.; Matsuura, K.; Haruta, S. Diversification of Bacterial Community Composition along a Temperature Gradient at a Thermal Spring. Microbes Environ. 2012, 27, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Hamamura, N.; Meneghin, J.; Reysenbach, A.-L. Comparative Community Gene Expression Analysis of Aquificales-Dominated Geothermal Springs. Environ. Microbiol. 2013, 15, 1226–1237. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MAG | Domain | Phylum | Class | Order | Family | Genus | Species | Method |
|---|---|---|---|---|---|---|---|---|
| 002 | Bacteria | Desulfobacterota | Thermodesulfobacteria | Thermodesulfobacteriales | Thermodesulfobacteriaceae | Caldimicrobium | Caldimicrobium sp009903935 | topology and ANI |
| 008 | Bacteria | Aquificota | Aquificae | Aquificales | Aquificaceae | Hydrogenobacter | - | topology and ANI |
| 010 | Bacteria | Thermotogota | Thermotogae | Thermotogales | Fervidobacteriaceae | Fervidobacterium | Fervidobacterium nodosum | topology and ANI |
| 014 | Bacteria | Dictyoglomota | Dictyoglomia | Dictyoglomales | Dictyoglomaceae | Dictyoglomus | Dictyoglomus thermophilum | topology and ANI |
| 016 | Bacteria | Spirochaetota | Leptospirae | Leptospirales | Leptonemataceae | - | - | RED |
| 019 | Bacteria | Deferribacterota | Deferribacteres | Deferribacterales | Calditerrivibrionaceae | Calditerrivibrio | - | RED |
| 023 | Bacteria | Aquificota | Aquificae | Hydrogenothermales | Hydrogenothermaceae | Sulfurihydrogenibium | Sulfurihydrogenibium azorense | topology and ANI |
| 026 | Bacteria | Aquificota | Aquificae | Aquificales | Aquificaceae | UBA11096 | UBA11096 sp011054805 | topology and ANI |
| 042 | Bacteria | Nitrospirota | Thermodesulfovibrionia | Thermodesulfovibrionales | Thermodesulfovibrionaceae | Thermodesulfovibrio | - | topology and ANI |
| 043 | Bacteria | Aquificota | Aquificae | Aquificales | Aquificaceae | - | - | topology |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escuder-Rodríguez, J.-J.; DeCastro, M.-E.; Saavedra-Bouza, A.; Becerra, M.; González-Siso, M.-I. Insights on Microbial Communities Inhabiting Non-Volcanic Hot Springs. Int. J. Mol. Sci. 2022, 23, 12241. https://doi.org/10.3390/ijms232012241
Escuder-Rodríguez J-J, DeCastro M-E, Saavedra-Bouza A, Becerra M, González-Siso M-I. Insights on Microbial Communities Inhabiting Non-Volcanic Hot Springs. International Journal of Molecular Sciences. 2022; 23(20):12241. https://doi.org/10.3390/ijms232012241
Chicago/Turabian StyleEscuder-Rodríguez, Juan-José, María-Eugenia DeCastro, Almudena Saavedra-Bouza, Manuel Becerra, and María-Isabel González-Siso. 2022. "Insights on Microbial Communities Inhabiting Non-Volcanic Hot Springs" International Journal of Molecular Sciences 23, no. 20: 12241. https://doi.org/10.3390/ijms232012241